主族元素光控配位數(shù):偶氮苯對(duì)主族元素取代基性質(zhì)的影響和它們的應(yīng)用
- 2012-08-13
- 專題
1、引述
每個(gè)主族元素在主族化合物通常都有它自己的特定的配位數(shù)、氧化數(shù)和粘結(jié)狀態(tài)。異常的化合價(jià)可以通過(guò)合適的分子設(shè)計(jì)和合成方法實(shí)現(xiàn),并且像這樣有異常化合價(jià)的元素具有獨(dú)特的特征、特殊的結(jié)構(gòu)和性質(zhì)。化合物的主要基團(tuán)的反應(yīng)能力明顯依賴于它們的配位數(shù)。例如,硅是14號(hào)元素,在周期表中位于碳元素的下方,通常它的配位數(shù)像sp3雜化的碳為4。硅也可以處于高配位數(shù)狀態(tài),例如在合適的條件下,硅在合適的配體中配位數(shù)為5或6。在高配位數(shù)的硅分子中,處于分子中心的硅原子具有比4配位數(shù)硅強(qiáng)的親電性,并且與硅原子相連的基團(tuán)的親核性也隨之增強(qiáng)(圖1)。因此,一些合成方法已經(jīng)被開(kāi)發(fā)通過(guò)利用高配位數(shù)的硅的性質(zhì),作為他們的中間體或過(guò)度狀態(tài),正如已被證實(shí)的Tamao氧化反應(yīng)中表現(xiàn)的性質(zhì)。1)另外其他例子如有機(jī)磷分子中的中心原子P顯三價(jià),它的配位數(shù)也為3,在過(guò)渡金屬中已廣泛應(yīng)用于配體。當(dāng)磷顯示五價(jià)并且四面體型配位或者顯示五價(jià)且配位數(shù)為5,磷的有效性將大大減少。
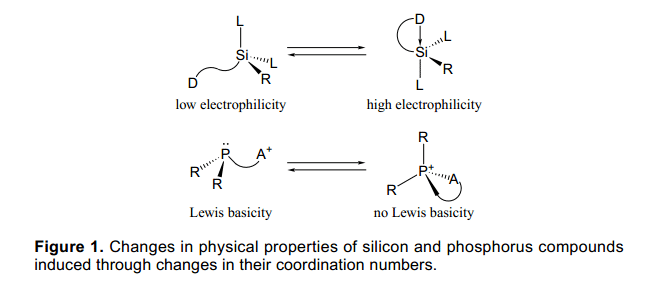
考慮到元素的性質(zhì)與配位數(shù)的關(guān)系、氧化態(tài)的控制、主族元素的配位數(shù),使用外部刺激很有意義。這將會(huì)產(chǎn)生新的方法通過(guò)控制物理性質(zhì)和分子中主要基團(tuán)性質(zhì)來(lái)控制有機(jī)反應(yīng)。例如,如果配位狀態(tài)很容易被改變,可以讓一個(gè)元素在特定階段表現(xiàn)一種處于激活狀態(tài)的配位數(shù),而在另一階段則表現(xiàn)為非激活狀態(tài),這將對(duì)共聚反應(yīng)規(guī)則非常有用。此外,如果這可以作為原料,它也可以作為一種新的保護(hù)方法或脫保護(hù)的配體。
然而,主族元素處于非常態(tài)的配位狀態(tài)通常是不穩(wěn)定的,而在實(shí)際應(yīng)用中有時(shí)是需要穩(wěn)定的狀態(tài)。例如,空間位阻常被用來(lái)來(lái)保護(hù)低配位數(shù)主族元素分子,通過(guò)在主族元素上引入體積較大的基團(tuán)以防止化合物的聚合。此外,處于高配位數(shù)狀態(tài)的元素被認(rèn)為容易發(fā)生消去反應(yīng),因?yàn)樗鼈冊(cè)谟袡C(jī)反應(yīng)中常是反應(yīng)過(guò)程中的中間體。因此,需要引入吸電子基團(tuán)來(lái)減弱中心原子的電子密度,并且增強(qiáng)極性鍵的穩(wěn)定性,多齒狀的配體應(yīng)該被用于有利的熵。在主族元素分子的非常態(tài)配位狀態(tài)下,分子穩(wěn)定性的設(shè)計(jì)中有許多限制因素。為了可逆的控制配位數(shù),控制中心原子與配體之間的關(guān)系顯得很重要。為了達(dá)到和這個(gè)目的,作為配體的取代基必須有這些滿足這些要求和可控性。因?yàn)檫@些可控性,不可能預(yù)先在主要組成化合物上開(kāi)發(fā)出特殊基團(tuán)來(lái)滿足接下來(lái)的兩點(diǎn)要求:1)穩(wěn)定的異常配位狀態(tài);2)結(jié)構(gòu)控制可以由外部因素激活。
一個(gè)可控的方法是通過(guò)外部因素激活:光照。已有幾種分子被證明他們的結(jié)構(gòu)隨著光照而改變。在它們當(dāng)中,偶氮苯作為光敏分子已被廣泛報(bào)道可以在各種類型的光照反應(yīng)中被光異性化。2)我們已經(jīng)將注意力定向在偶氮苯的這個(gè)性質(zhì)上,也就是,偶氮苯的一種結(jié)構(gòu)具備一定的剛度并能感光異構(gòu),以及偶氮基團(tuán)可以起到路易斯堿或利用試劑作為親電體。我們希望當(dāng)偶氮苯作為分子內(nèi)配體時(shí),可以在(E)—和(Z)—之間可逆的光異構(gòu)化來(lái)控制主族元素的配位數(shù)。首先,我們?cè)谂嫉降?號(hào)位引入一個(gè)主族元素基團(tuán),因此,我們可以利用偶氮基團(tuán)作為光敏點(diǎn)、配位點(diǎn)或親電點(diǎn)。我們實(shí)驗(yàn)的結(jié)果是主族元素的碳、硅、磷的配位數(shù)在光照條件下可以控制,并且他們的結(jié)構(gòu)、物理性質(zhì)和反應(yīng)活性在幾種體系中均可很好的光控。在這篇論文中,我們主要關(guān)注這幾種方法的發(fā)展、介紹幾種控制主族元素配位數(shù)的控制方法以及他們的應(yīng)用,我們也討論了偶氮苯自身作為取代基的性質(zhì)。
2.硅烷和硅酸鹽的光控結(jié)構(gòu)以硅元素的配位數(shù)為基礎(chǔ)
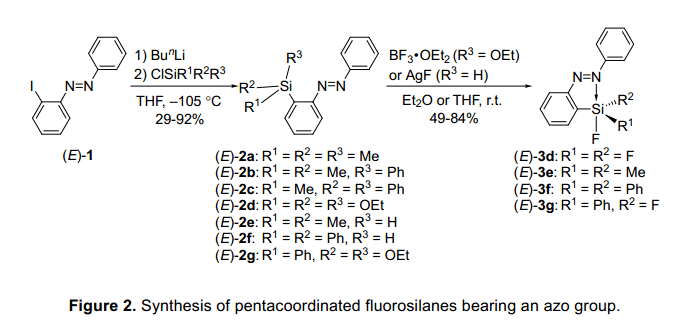
一個(gè)2-(苯偶氮基)苯基引入到硅元素上是通過(guò)一個(gè)有機(jī)鋰衍生物,它的制備是在-105°C的條件下,2-碘基偶氮苯(E)-1在丁基鋰的作用下進(jìn)行的(如圖2)。3,4)硅的配位態(tài)是很容易通過(guò)29Si的核磁共振波譜確認(rèn)的。2 -(苯偶氮基)苯基衍生物和與此相似的苯基衍生物的化學(xué)位移的差異只有±2ppm,這表明硅元素提供了一個(gè)4配位態(tài),其偶氮基團(tuán)的配位態(tài)幾乎沒(méi)有變化。因此,期望硅的親電性通過(guò)氟化作用會(huì)增加,單氟硅烷(E)-3e和(E)-3f的制備是通過(guò)單氫硅烷(E)-2e 和(E)-2f分別與氟化銀反應(yīng)的。雙乙氧基硅烷(E)-2g和三乙氧基硅烷(E)-2d分別與BF3?OEt2反應(yīng)制取雙氟硅烷(E)-3g和三氟硅烷(E)-3d。(E)-3d-g中硅元素的配位態(tài)與類似的苯基衍生物通過(guò)29Si的核磁共振來(lái)相比較,就會(huì)發(fā)現(xiàn)(E)-3d-g的化學(xué)位移高出11 ppm多。科學(xué)家發(fā)現(xiàn)硅原子的5配位態(tài)通過(guò)偶碳基團(tuán)協(xié)調(diào)作用,氟硅烷衍生物在溶液明顯表現(xiàn)出光譜屬性。是否存在這種配位作用是經(jīng)x-線衍射單晶分析確認(rèn)的 。若化合物中沒(méi)有氟原子,體積龐大的硅取代基是被發(fā)現(xiàn)位于氮原子的對(duì)位。通過(guò)對(duì)比,結(jié)果表明,在硅原子上存在至少一個(gè)氟原子,五配位態(tài)的硅原子可以與一對(duì)孤對(duì)氮原子相互作用,導(dǎo)致硅原子周圍的的周圍形成一個(gè)扭曲的三方雙錐體結(jié)構(gòu)。形成一個(gè)N-Si配鍵誘導(dǎo)代換氟原子是歸因于增加親電性的硅原子和電負(fù)性氟原子,正如在前面報(bào)道的,有機(jī)硅烷的配體數(shù)不同于偶氮基團(tuán)。
硅原子不同的配位狀態(tài)可以通過(guò)化合物的晶體顏色很容易地區(qū)別出來(lái)。四配位態(tài)的含硅偶氮苯且沒(méi)有氟原子,顯示一個(gè)紅色或橙色的色彩,五配位態(tài)的含硅偶氮苯(E)-3d-g顯示黃色。通過(guò)紫外/可見(jiàn)光譜觀察在二氯甲烷的溶液里的四配位態(tài)(E)-2a-g化合物,他們的吸收最大值歸結(jié)為p-p* 和n-p*,分別轉(zhuǎn)換為325-327nm和441-456nm。與沒(méi)有取代基的偶氮苯(p-p* 318nm ,n-p* 轉(zhuǎn)換為441nm)相比,四配位態(tài)(E)-2a-g化合物略紅移,這種紅移證明通過(guò)有機(jī)硅基團(tuán)的供電子基的誘導(dǎo)效應(yīng)提高了n-和p-軌道的能級(jí)。相比之下,在偶氮苯(E)-3d-g與五配位態(tài)的硅取代基,最大吸收值歸結(jié)為p-p*躍遷觀察到的紅移約336-349nm,而最大吸收值歸結(jié)為n-p*躍遷觀察到相對(duì)藍(lán)光偏移。因此,不容易發(fā)現(xiàn)化合物(E)-3d或者(E)-3g存在n-p *躍遷吸收光譜,可能是因?yàn)檫@是藏在p-p*躍遷。這些結(jié)果表明,氮原子和硅原子形成一個(gè)配位鍵穩(wěn)定了n軌道并且π軌道變得比π*軌道相對(duì)不穩(wěn)定。
當(dāng)(E)-3d-g用波長(zhǎng)為p-p*躍遷(l=360nm)的紫外燈照射時(shí),使用超高壓水銀燈和一個(gè)彩色的玻璃過(guò)濾器,他們異構(gòu)化成相應(yīng)的化合物(Z)-3d-g,并且具有81~99%的高收益率(如圖3)。異構(gòu)化作用后,產(chǎn)生這些化合物顯示橙色。最大吸收值在大約340nm處下降,與此同時(shí)在440nm處有一個(gè)最大吸收值,這是因?yàn)閜-p*躍遷增加了。此外,當(dāng)(Z)-異構(gòu)體用波長(zhǎng)為l=431nm紫外燈照射,(Z)-異構(gòu)體能被可逆地轉(zhuǎn)變?yōu)?E)-異構(gòu)體。
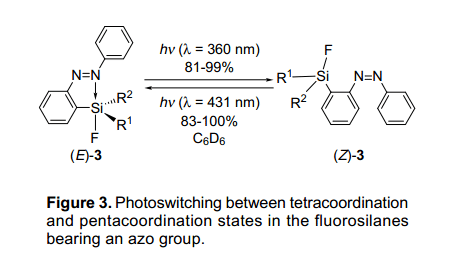
(Z)-異構(gòu)體在光異構(gòu)化作用后的配位態(tài)可以通過(guò)Si NMR檢測(cè)。(Z)-同分異構(gòu)體與相應(yīng)的(E)-同分異構(gòu)體相比其化學(xué)位移要低些,但和苯基衍生物的化學(xué)位移相似,這說(shuō)明(Z)—同分異構(gòu)體具有四配位態(tài)。特別是,氟代甲硅烷基聯(lián)苯衍生物的兩個(gè)(E)-3f 和(Z)-3f同分異構(gòu)體可能被孤立,這表明同分異構(gòu)體(E)-3f和(Z)-3f的硅原子分別具有五配位態(tài)和四配位態(tài)。這兩種結(jié)構(gòu)通過(guò)x-射線衍射單晶分析被進(jìn)一步證實(shí)了,通過(guò)光異構(gòu)化進(jìn)一步展示了硅原子的結(jié)構(gòu)變化。
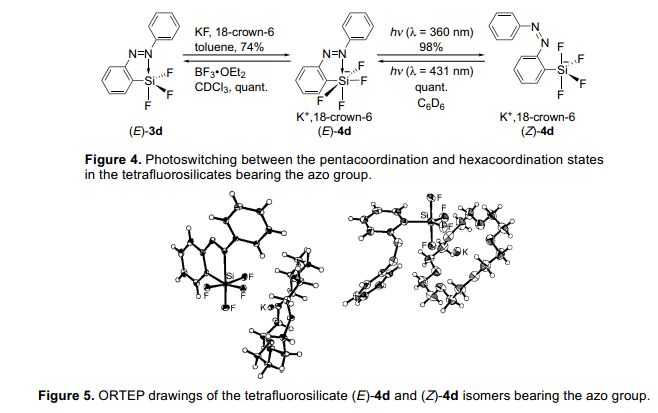
由于我們成功地光轉(zhuǎn)化氟代硅烷的五配位態(tài)和四配位態(tài),我們下一步試圖通過(guò)類似的方法用不同配位態(tài)的硅原子開(kāi)發(fā)各種光轉(zhuǎn)化條件。緊接著是五配位態(tài)的研究,下一步是研究六配位態(tài)。一個(gè)硅酸鹽和分子配位很容易形成六配位態(tài)。我們?cè)噲D進(jìn)一步產(chǎn)生更高的配位態(tài)的硅原子通過(guò)把(E)-3d轉(zhuǎn)換成硅酸鹽。我們分析三氟硅烷(E)-3d的氟化作用通過(guò)添加氟化鉀和18-冠-6醚和已制取的硅酸鹽(E)-4d(圖4)。5)當(dāng)用波長(zhǎng)類似為偶氮基團(tuán)的p-p*躍遷的紫外燈照射黃色硅酸鹽(E)-4d時(shí),異構(gòu)化幾乎定量進(jìn)行,形成了紅橙色硅酸鹽(Z)-4d。異構(gòu)化的(Z)-4d異構(gòu)體也定量轉(zhuǎn)變成(E)-4d異構(gòu)體,暗示(E)-和 (Z)-同分異構(gòu)體可能定量的進(jìn)行光異構(gòu)化。低溫條件下19F和29Si核磁共振光譜和x-射線衍射單晶分析表明(E)-4d的六配位態(tài)的硅原子有一個(gè)扭曲的八面體結(jié)構(gòu),盡管(Z)-4d五配位態(tài)硅原子顯示一個(gè)三角形的雙錐體結(jié)構(gòu)(圖5)。最戲劇性的變結(jié)構(gòu)化在被發(fā)現(xiàn)在F-Si-F角度,這E)-4d異構(gòu)體的角度是99.47(5)°和163.73(6)°,雖然(Z)-4d異構(gòu)體的鍵角度分別是117.77(19)°和126.67(19)°。此外,(E)-4d,的F-Si-F鍵角是95.76(5)變?yōu)?15.53(6)°。這是值得注意的是,光致輻照獨(dú)自誘導(dǎo)這樣一個(gè)偉大的結(jié)構(gòu)變化并且在硅原子的周圍沒(méi)有添加任何化學(xué)試劑。因此,我們已經(jīng)成功地通過(guò)光致輻照五和六配位態(tài)硅酸鹽4d控制配位數(shù)變化,這一直伴隨控制硅原子周圍的構(gòu)象。
有許多的報(bào)告中,化合物物理屬性的光轉(zhuǎn)化是通過(guò)使用偶氮苯的光異構(gòu)化。在這之前研究,在大多數(shù)情況下取代基是位于偶氮基的對(duì)位或間位,2)通過(guò)調(diào)(E)和(Z)同分異構(gòu)體的每個(gè)取代基位置關(guān)系或方向,尋找光控條件獲得化合物的物理性質(zhì)。與這些以前光轉(zhuǎn)化系統(tǒng)不同,然而,我們?cè)谶@里介紹的光轉(zhuǎn)化系統(tǒng)是獨(dú)特的,硅原子結(jié)構(gòu)可以交換使用在硅和孤對(duì)氮原子之間的位置關(guān)系的變化。即使化合物的有機(jī)軸承取代基的硅原子,如果取代基的方向可以交換通過(guò)使用這個(gè)方法并且這合成的物質(zhì)進(jìn)一步結(jié)合定向調(diào)控的偶基團(tuán)的對(duì)位取代基,這個(gè)系統(tǒng)可能會(huì)應(yīng)用于各種類型物理性質(zhì)的光轉(zhuǎn)變條件。
3.二氧化硅基于硅原子的配位數(shù)變化而導(dǎo)致光轉(zhuǎn)換活性的變化
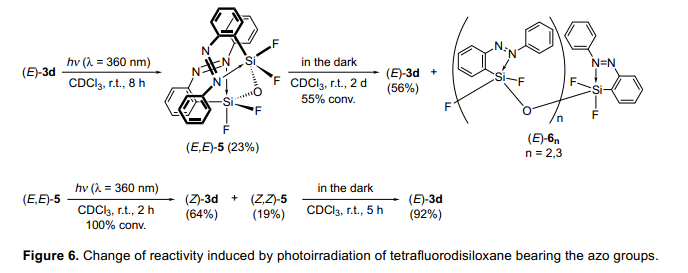
在前面兩部分中,我們已經(jīng)介紹光照可以控制含硅分子的生成結(jié)構(gòu)。那么,接下來(lái)的問(wèn)題已經(jīng)出現(xiàn),“我們能否控制他們的反應(yīng)活性?”當(dāng)(E)-3d三氟硅烷的穩(wěn)定性被測(cè)定后,研究發(fā)現(xiàn)(E)-3d三氟硅烷在(E,E)-5-1,1,3,3,—四氟二甲硅醚漸漸水解了。(圖6)(E,E)-5在其分子內(nèi)連接了兩分子的偶氮苯。考慮到偶氮苯可以輕松的堆疊成結(jié)晶狀態(tài),兩分子偶氮苯利用Si-O-Si之間的間隔可以并排在兩個(gè)苯環(huán)的3.4—3.6?空間內(nèi)。在避光條件室溫下,(E,E)-5二甲硅烷醚在兩天內(nèi)進(jìn)一步降解,形成(E)3d和(E)-6n(n=2,3)低聚硅氧烷的混合物。光照可以催化(E,E)-5的降解,并且光照2h后,(E,E)-5全部降解,由于這個(gè)反應(yīng),四配位數(shù)的(Z,Z)-5二甲硅氧醚的兩個(gè)偶氮基團(tuán)被異構(gòu)化,除了生成(Z)-3d。當(dāng)(E,E)-5在避光室溫條件下將生成(E)-3d,并且由(Z,Z)-5的合成速率比(E,E)-5的大。
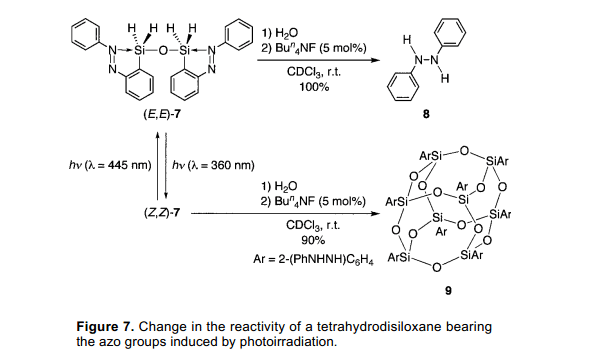
對(duì)于(E,E)-7分子,相應(yīng)的(E,E)-5的四氫衍生物的反應(yīng)活性在光照前后有明顯的變化。在一定量的四丁胺氟化物催化下,(E,E)-7被水解成二苯肼8伴隨著C-Si鍵的斷裂。(圖7)7)另外,當(dāng)嘗試類似的方法使用四配位數(shù)的硅化物(E,E)-7可以定量的光照(Z,Z)-7制得。倍半硅氧烷9的制備沒(méi)有C-Si鍵的斷裂,盡管偶氮基團(tuán)減少了。因此,反應(yīng)活性的改變伴隨著C-Si鍵的斷裂可以被光照條件控制。
4.光變對(duì)丙烯硅烷反應(yīng)活性的影響:改變硅原子的配位數(shù)
正如在前面章節(jié)中展示的,眾周所知,有機(jī)硅分子的活性因其配位數(shù)的不同而明顯不同。盡管這里在我們沒(méi)有詳細(xì)討論它們的反應(yīng)機(jī)理,不同的活性伴隨不同的配位數(shù)有如下原因。相比于電荷形式,硅與取代基鍵顯示極性,三中心四電子鍵存在于五或六配位數(shù)狀態(tài)。這將導(dǎo)致取代基親電性的增強(qiáng),并且取代基活性也比四配位狀態(tài)強(qiáng)。一般來(lái)說(shuō),硅原子的配位數(shù)可以由四配位狀態(tài)改良為更高的五或六配位狀態(tài),通常是增加一種試劑到反應(yīng)體系來(lái)產(chǎn)生一個(gè)高活性的配位物質(zhì)。然而,正如上面所描述的,控制配位數(shù)很容易實(shí)現(xiàn),只需要光照偶氮基團(tuán)而不需要添加其他任何化學(xué)試劑。下一步,我們嘗試著控制Hosomi-Sakura反應(yīng),這是一個(gè)著名的利用硅試劑的反應(yīng),并研究光控烯丙硅烷的活性對(duì)偶氮基的影響。
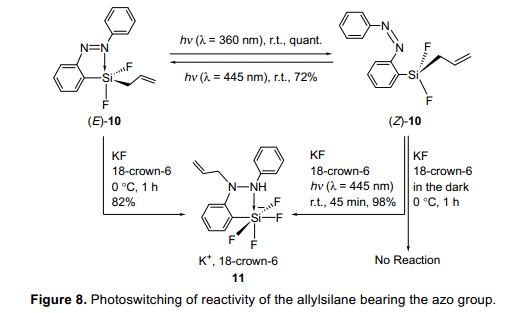
具有偶氮基團(tuán)的烯丙基雙氟代硅烷(E)-10被合成,其協(xié)配位態(tài)通過(guò)使用多核磁共振光譜和x-射線衍射單晶分析被研究。正如預(yù)期的那樣,這是證明(E)-10的硅原子顯示五配位態(tài),而光異構(gòu)化得到的(Z)-10的硅原子顯示四配位態(tài)。苯甲醛新增了一項(xiàng)親電基,但是沒(méi)有反應(yīng)的進(jìn)行。因此,在18-冠-6醚存在的條件下,氟化鉀與(E)-10反應(yīng)是為了激活(E)-10,結(jié)果導(dǎo)致N-烯丙基硅酸鹽11(N-allylated silicate 11)的產(chǎn)生(圖8)。另一方面,緊接著(E)-10被光異構(gòu)化成(Z)-10,其次是在黑暗條件下氟化鉀存在于18-crown-6沒(méi)有反應(yīng)。有趣的是,光致輻照誘導(dǎo)了反應(yīng)繼續(xù)并生成化合物11,類似的例子(E)-10。由于在光致輻照條件下,五配位態(tài)的(E)-10和四配位態(tài)的(Z)-10可能是相互的轉(zhuǎn)化的;這些結(jié)果表明光致輻照條件可以控制烯丙基硅烷的反應(yīng)性。結(jié)果從幾個(gè)控制的實(shí)驗(yàn)顯示從(E)-10形成化合物11是一個(gè)分子內(nèi)反應(yīng),在這個(gè)反應(yīng)中,烯丙基基團(tuán)從硅原子轉(zhuǎn)移到氮原子上在g位。這說(shuō)明,氮配位誘導(dǎo)增加烯丙基基團(tuán)的親核性,也增加了硅原子的親電性,增強(qiáng)偶氮基團(tuán)的極化并且六元環(huán)過(guò)渡態(tài)都在這些反應(yīng)的過(guò)程扮演著一個(gè)重要的角色。
當(dāng)偶氮集團(tuán)被認(rèn)為是一種反應(yīng)底物,(Z)-和(E)-同分異構(gòu)體可以分別被認(rèn)為對(duì)應(yīng)被保護(hù)和脫保護(hù)的基團(tuán),這適用于多步反應(yīng)包括上面描述的烯丙基化反應(yīng)。換句話說(shuō),保護(hù)和脫保護(hù)的反應(yīng)基團(tuán)是通過(guò)使用不同波長(zhǎng)的光輻射。
5.硼化合物的劉易斯酸光轉(zhuǎn)化攻擊偶氮基團(tuán)
作為主族元素的一個(gè)實(shí)際應(yīng)用,除了增加取代基的親核性,有其他一些方法可以修改主族元素本身的屬性,譬如硅化合物。例如,有機(jī)硼化合物具有劉易斯酸的性能和該性能已被廣泛應(yīng)用于各種有機(jī)合成反應(yīng)。作為一個(gè)基本研究控制與硼化合物反應(yīng)的反應(yīng)性例如路易斯酸催化劑,我們調(diào)查了光是否控制配位數(shù),兒茶酚硼烷攻擊偶氮苯的一部分及其劉易斯酸,評(píng)價(jià)劉易斯酸的變化使用與吡啶生成絡(luò)合物作為其反應(yīng)指數(shù)。
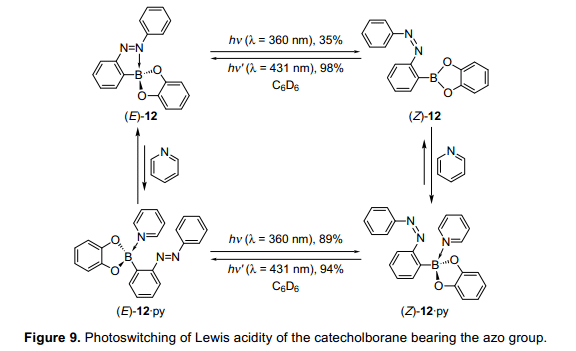
硼酸與偶氮苯基團(tuán)發(fā)生脫水縮合反應(yīng),生成兒茶酚硼烷(E)-12。光致輻照條件下,(E)-12在苯溶液中可逆地進(jìn)行光異構(gòu)化,盡管其異構(gòu)化效率不高(圖9)。等摩爾數(shù)的吡啶與兒茶酚硼烷(Z)-12反應(yīng),11B核磁共振的結(jié)果顯示在前場(chǎng)的明顯轉(zhuǎn)變,這是證明了一個(gè)復(fù)雜的成立,吡啶作為一個(gè)路易斯堿存在這種異構(gòu)體。相比之下,當(dāng)(E)-12的異構(gòu)體與吡啶反應(yīng),化學(xué)位移沒(méi)有顯著的變化,表明(E)-12的分子內(nèi)配位可能會(huì)擾亂吡啶分子間配位,導(dǎo)致(E)-12與吡啶不能形成絡(luò)合物。此外,光異構(gòu)化反應(yīng)可逆地進(jìn)行,甚至在吡啶過(guò)量的條件下。即,當(dāng)用紫外燈照射(Z)-12?py時(shí),(E)-12?py和(E)-12之間的平衡是明顯轉(zhuǎn)向(E)-12的。這些結(jié)果顯示,大多數(shù)吡啶分子形成復(fù)合物而兒茶酚硼烷可能被釋放硼原子和變得自由。當(dāng)改變等摩爾數(shù)吡啶后,通過(guò)11B核磁共振估計(jì)化學(xué)位移值,來(lái)測(cè)量絡(luò)合物的絡(luò)合值。我們發(fā)現(xiàn),絡(luò)合物增加了大約300倍,通過(guò)光異構(gòu)化使(E)-12轉(zhuǎn)變?yōu)?Z)-12。因此,由結(jié)果判斷與吡啶分子形成絡(luò)合物,通過(guò)劉易斯酸在硼化合物被成功執(zhí)行。
6.膦基偶氮苯和分子內(nèi)鏻鹽之間的平衡控制
在上面所描述的硅或硼化合物,偶氮基團(tuán)在這些化合物中擔(dān)任親核試劑的作用。然后我們考慮是否有可能控制主族元素的配位數(shù),當(dāng)偶氮基團(tuán)也作為一個(gè)親電試劑。有機(jī)磷化合物是一個(gè)主族化合物作為一個(gè)親核試劑或者一個(gè)路易斯堿在一個(gè)正常的配位態(tài)下。例如,磷化氫已經(jīng)被廣泛的用作與過(guò)渡金屬形成配體。如果我們能表明我們可以控制有機(jī)磷化合物的配位數(shù),那是可以預(yù)期,我們也許能夠控制化合物的配位能力通過(guò)過(guò)渡金屬和由他們合成的催化劑活性。
2-鋰基偶氮苯被二苯氯化磷接受并且膦類化合物的合成嘗試通過(guò)類似反應(yīng)使用硅和硼。然而,沒(méi)有獲得相應(yīng)的膦類化合物。經(jīng)過(guò)反復(fù)嘗試的過(guò)程,我們發(fā)現(xiàn)2-碘基偶氮苯(E)-1被轉(zhuǎn)換成磷氫基硫化物(E)-14通過(guò)與二苯肼(E)-13反應(yīng)。(E)-14與三丁基磷化氫經(jīng)過(guò)脫硫作用導(dǎo)致2-雙苯基膦基偶氮苯(E)-15的合成(圖10)。12)化合物(E)-14也可以得到通過(guò)2-鋰基偶氮苯與雙苯基硫代磷酸化異煙酸氯化物(13)的反應(yīng)。自膦基基團(tuán)通常作為一個(gè)親核試劑,但不是一個(gè)親電體,估計(jì)偶氮基團(tuán)可能作為一個(gè)電子受體。然而,化合物(E)-15顯示一個(gè)磷化氫結(jié)構(gòu),其中在晶體狀態(tài)中偶氮基團(tuán)沒(méi)有交互氮和磷原子。化合物(E)-15在甲苯溶液中顯示熱致變色和在100°C下顯示一個(gè)紅色的顏色,但在低溫條件如-78°C時(shí)變成黑色,這樣的情況和通常的偶氮苯是不同的。此外,化合物(E)-15的31P核磁共振化學(xué)位移表現(xiàn)出明顯的溫度依賴性。在溫度高于60°C時(shí),它在-9.2ppm顯示一個(gè)單線態(tài),在20°C和-60 °C之間顯示一個(gè)廣泛的信號(hào),并且低于-60°C時(shí)在30.1 ppm和-10.3 ppm處呈單譜線。在強(qiáng)磁場(chǎng)信號(hào)屬于膦結(jié)構(gòu),而信號(hào)低場(chǎng)屬于分子內(nèi)磷鹽16基于比較觀測(cè)到的化學(xué)變化和這些估計(jì)的GIAO計(jì)算。這表明分子內(nèi)磷鹽16應(yīng)該是分子內(nèi)磷化氫基團(tuán)攻擊偶氮基團(tuán)產(chǎn)生的物質(zhì)。黑顏色是在較低的溫度下對(duì)應(yīng)的陰陽(yáng)離子16的過(guò)渡分子內(nèi)電荷轉(zhuǎn)移的,而紅顏色是歸因于在高溫度下偶氮基團(tuán)的p-p*躍遷。
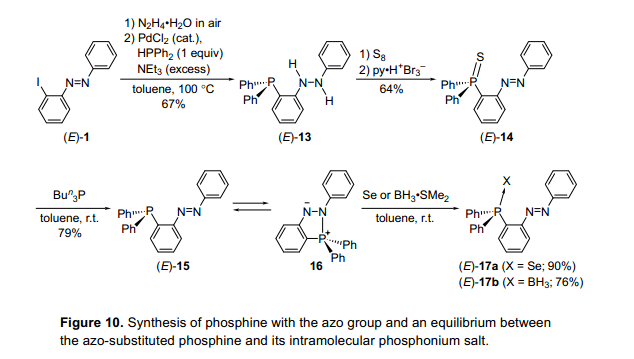
考慮到所有這些結(jié)果,在室溫下磷化氫化合物(E)-15和分子內(nèi)磷鹽16形成一個(gè)平衡。 當(dāng)溫度升高,平衡是轉(zhuǎn)向(E)-15,而低溫度導(dǎo)致轉(zhuǎn)向化合物16。這證明了我們可以成功地通過(guò)改變溫度控制膦(E)-15和磷鹽16的平衡。當(dāng)反應(yīng)在一個(gè)平衡溶液是使用是與硒B(niǎo)H3?SMe2、磷化氫硒化物(E)-17a和磷化氫硼烷(E)-17b反應(yīng)形成,表明這個(gè)磷化氫有普遍的反應(yīng)性。它可以很容易地與水反應(yīng)而不像往常膦類化合物。例如,類似的衍生物(E)-15’在4位有一個(gè)甲基,存在一個(gè)很少量的水促進(jìn)磷化氫氧化物18的聯(lián)苯肼基的形成。分子內(nèi)磷鹽來(lái)源于2-膦基偶氮苯,顯示與Mitsunobu反應(yīng)的一個(gè)中間體相似的性能。14)它們的反應(yīng)性也支持膦類化合物和分子內(nèi)季磷鹽存在一個(gè)平衡。
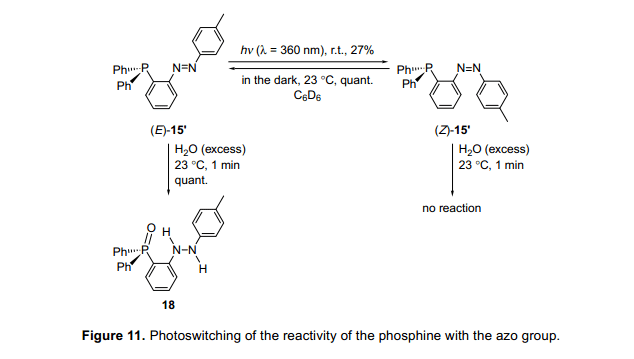
然后,我們懷疑這些膦化合物的配位數(shù)可能受光致輻照的控制,光致輻照(E)-15發(fā)并現(xiàn)沒(méi)有異構(gòu)化發(fā)生。然而,有一個(gè)甲基(E)-15''經(jīng)光輻射,光異構(gòu)化后產(chǎn)生(Z)-15”(圖11)。13)然后我們監(jiān)控(E)-15”或(Z)-15”與水的反應(yīng)。(E)-15'與水迅速反應(yīng),一分鐘后完全消失,盡管沒(méi)有觀察到(Z)-15'的變化。最后,在很長(zhǎng)的一段時(shí)間的反應(yīng)之后(Z)-15”也產(chǎn)生了18,這是由于它的熱異構(gòu)化。因此,磷化氫的反應(yīng)性能是在一個(gè)有限的時(shí)間內(nèi)可以控制光致輻照分子內(nèi)磷鹽向水均衡。
7.開(kāi)發(fā)的熒光偶氮苯與硼取代基
在前面的章節(jié)中討論的一樣,在2位置含有主族元素取代基的偶氮苯顯示各種各樣的顏色如黃色、橙色、紅色和黑色。實(shí)際上,通過(guò)修改偶氮苯取代基,偶氮苯可以進(jìn)一步顯示各種不同的顏色,并且偶氮苯的這個(gè)性能已被用作偶氮染料的基本結(jié)構(gòu)性能。偶氮染料幾乎占了全球一半的工業(yè)染料生產(chǎn),并且在印染行業(yè)中占據(jù)著重要的地位。一些顏料,它被稱為熒光染料,不用吸收一個(gè)特定光譜的光線,但也能發(fā)出光作為熒光。然而,在室溫下,偶氮苯一般不會(huì)發(fā)出任何重要的熒光產(chǎn)生光致輻照,因?yàn)楣猱悩?gòu)化通常在偶氮苯中是收益的。據(jù)報(bào)道,一些含鈀化合物與偶氮苯作為一個(gè)配體15)和一些聚合體16)攻擊偶氮苯,作為一個(gè)組件可以異常發(fā)出弱熒光。這含有偶氮苯的鈀配合物作為配體,然而,熒光量子收益率只有10-5到10-3。
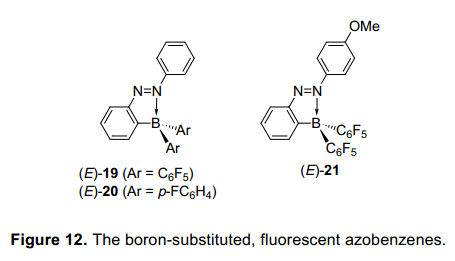
我們已經(jīng)合成偶氮苯的化合物,這可以接受光異構(gòu)化,而不受互動(dòng)的含氮原子的偶氮基團(tuán)和主族元素的限制。對(duì)于含有2—硼取代基的偶氮苯的異構(gòu)化產(chǎn)量與硅相比很低。這是需要找到一種硼取代基偶氮苯,可以更有效地在光致輻照條件下異構(gòu)化。在這個(gè)隨后的研究中,我們調(diào)查了大量含偶氮苯基團(tuán)或硼原子的芳環(huán)上不同的取代基,并檢查了取代基之間的關(guān)系類型和他們的光學(xué)性質(zhì)。因此,我們發(fā)現(xiàn)一個(gè)平衡在三配位態(tài)和四配位態(tài),傾向于轉(zhuǎn)向四配位態(tài),當(dāng)有一個(gè)化合物有供電子偶氮苯基團(tuán)的4位上或一個(gè)吸電子基在硼原子上,這將導(dǎo)致增加可吸收光譜。此外,它也表明了他們配價(jià)鍵增強(qiáng),異構(gòu)化率降低,并且有一些化合物未能進(jìn)行異構(gòu)化。這些結(jié)果表明,硼取代偶氮苯化合物不適合配位數(shù)的控制,這也成為我們最重要的目標(biāo)。然而進(jìn)一步詳細(xì)分析了,表明在各種硼取代基的偶氮苯基團(tuán)不能光異構(gòu)化,在室內(nèi)光線下2(五氟苯酚)-硼基-偶氮苯(E)-19在溶液中發(fā)出綠色熒光(圖12)。17)在室溫下在己烷溶液中,熒光量子產(chǎn)率為0.23,這相對(duì)偶氮苯是較高的。然而,化合物(E)-20的五氟苯酚基團(tuán)被對(duì)氟苯酚基團(tuán)所取代,但是不發(fā)射任何熒光。這些結(jié)果表明,N-B共價(jià)鍵的鍵能和熒光強(qiáng)度之間可能有關(guān)系。因此,我們接下來(lái)調(diào)查了芳環(huán)上的不同取代基,發(fā)現(xiàn)化合物(E)-21有一個(gè)甲氧基在偶氮苯的4位上具有更強(qiáng)的熒光,并且在室溫下己烷溶液中它的量子產(chǎn)率是0.76。在曾被報(bào)道偶氮苯衍生物中,這個(gè)熒光量子產(chǎn)率值是最高的。它表明,(E)-21發(fā)射熒光比未被取代的偶氮苯發(fā)射的30000倍還強(qiáng)。
因此,我們下一步試圖從理論和實(shí)驗(yàn)上確定硼取代偶氮苯化合物發(fā)射的熒光原因。這是推測(cè),N-B配位鍵可能不會(huì)只抑制偶氮苯的結(jié)構(gòu)變化,也相對(duì)提高p軌道的能級(jí)通過(guò)降低n軌道的能級(jí)。這倒逆轉(zhuǎn)變化能級(jí)會(huì)使p-p*躍遷從基態(tài)躍遷到到最低的能級(jí)。因此,從最低的能級(jí)躍遷到基態(tài)是容易發(fā)生的,這進(jìn)一步導(dǎo)致在一個(gè)高產(chǎn)率下吸收光能產(chǎn)生熒光。在最開(kāi)始的研究中,我們沒(méi)有設(shè)想,引入硼取代基到偶氮苯會(huì)誘導(dǎo)熒光發(fā)射。然而,這里所介紹的硼取代偶氮苯比報(bào)道鈀配合物發(fā)出更強(qiáng)烈的熒光。主族元素獨(dú)特的性質(zhì)提供了偶氮苯的更強(qiáng)烈的熒光,而沒(méi)有使用昂貴的鈀。
8.總結(jié)
我們介紹了一個(gè)模型系統(tǒng),可以變換主族化合物的物理性質(zhì)和結(jié)構(gòu),通過(guò)使用光致輻照誘導(dǎo)偶氮苯的異構(gòu)化作用,作為一個(gè)關(guān)鍵的反應(yīng)。這種方法同樣被認(rèn)為是適用于其他主族元素,它被高度期望動(dòng)態(tài)地控制元素特定的物理性質(zhì)而不僅僅是以上描述的那些。如果我們能進(jìn)一步開(kāi)發(fā)新的方法,可以直接控制主族化合物的配位數(shù),并基于上述闡述的概念,我們相信這種方法的應(yīng)用將打開(kāi)一個(gè)新方法控制有機(jī)反應(yīng),這激活/失活方法的使用類似主族元素的配位態(tài)的方法。最近報(bào)道,一個(gè)新的有效合成方法已開(kāi)發(fā)使用,掩蔽和暴露硼元素,18)對(duì)于這樣的目的如果這里介紹的方法能夠得到應(yīng)用,這個(gè)新方法它是可行的。當(dāng)前的研究結(jié)果的理論和實(shí)驗(yàn),并且等同于實(shí)際使用和應(yīng)用在一個(gè)在實(shí)踐層面上。我們認(rèn)為,然而,我們?cè)谶@里的概念與傳統(tǒng)的是不同的。因此,我們將會(huì)很高興如果這個(gè)獨(dú)特的概念可以用來(lái)控制有機(jī)反應(yīng)或化學(xué)過(guò)程,如保護(hù)胺。非常值得一提的是,在研究主族化合物時(shí),我們發(fā)現(xiàn)一個(gè)意想不到的,令人驚訝的產(chǎn)品——熒光偶氮苯。我們確信主族化合物一定存在很大的研究?jī)r(jià)值,等待我們?nèi)グl(fā)現(xiàn)和利用。
參考文獻(xiàn)
1. (a) Tamao, K.; Ishida, N.; Tanaka, T.; Kumada, M.Organometallics 1983, 2, 1694. (b) Tamao, K., J. Synth.Org. Chem. Japan, 1988, 46, 861.
2. (a) Molecular Switches; Feringa, B. L., Ed.; Wiley-VCH:Weinheim, 2001. (b) Balzani, V.; Credi, A.; Raymo, F.M.; Stoddart, J. F. Angew. Chem., Int. Ed. 2000, 39,3348. (c) Feringa, B. L.; van Delden, R. A.; Koumura,N.; Geertsema, E. M. Chem. Rev. 2000, 100, 1789. (d)Ichimura, K. Chem. Rev. 2000, 100, 1847.
3. Kano, N.; Komatsu, F.; Kawashima, T. Chem. Lett.2001, 338.
4. Kano, N.; Komatsu, F.; Yamamura, M.; Kawashima, T.J. Am. Chem. Soc. 2006, 128, 7097.
5. Kano, N.; Komatsu, F.; Kawashima, T. J. Am. Chem.Soc. 2001, 123, 10778.
6. Kano, N.; Yamamura, M.; Komatsu, F.; Kawashima, T.J. Organomet. Chem. 2003, 686, 192.
7. Yamamura, M.; Kano, N.; Kawashima, T. TetrahedronLett. 2007, 48, 4033.
8. (a) Hosomi, A.; Sakurai, H. Tetrahedron Lett. 1976,1295. (b) Hosomi, A. Acc. Chem. Res. 1988, 21, 200.
9. Kano, N.; Yamamura, M.; Kawashima, T. J. Am. Chem.Soc. 2004, 126, 6250.
10. Yamamura, M.; Kano, N.; Kawashima, T. J. Organomet.Chem. 2007, 692, 313.
11. Kano, N.; Yoshino, J.; Kawashima, T. Org. Lett. 2005,7, 3909.
12. Yamamura, M.; Kano, N.; Kawashima, T. Inorg. Chem.2007, 45, 6497.
13. Yamamura, M.; Kano, N.; Kawashima, T. J. Am. Chem.Soc. 2005, 127, 11954.
14. (a) Mitsunobu, O. Synthesis 1981, 1, 1. (b) Hughes, D.L. Organic Reactions; Wiley: New York, 1992; Vol. 42,pp 335-656.
15. (a) Wakatsuki, Y.; Yamazaki, H.; Grutsch, P. A.;Santhanam, M.; Kutal, C. J. Am. Chem. Soc. 1985,107, 8153. (b) Ghedini, M.; Pucci, D.; Crispini, A.; Aiello,I.; Barigelletti, F.; Gessi, A.; Francescangeli, O. Appl.
Organomet. Chem. 1999, 13, 565.
16. (a) Shimomura, M.; Kunitake, T. J. Am. Chem. Soc.1987, 109, 5175. (b) Han, M.; Hara, M. J. Am. Chem.Soc. 2005, 127, 10951.
17. Yoshino, J.; Kano, N.; Kawashima, T. Chem. Commun.2007, 559.
18. Noguchi, H.; Hojo, K.; Suginome M. J. Am. Chem. Soc.1985, 107, 8153.
注:本文為提供者整理翻譯的,由于知識(shí)所限,錯(cuò)誤在所難免,敬請(qǐng)?jiān)彙H缬袉?wèn)題可以查找原文。
統(tǒng)計(jì)數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)
-
物竟數(shù)據(jù)庫(kù) 手機(jī)版
國(guó)內(nèi)首款化工類專業(yè)手機(jī)應(yīng)用
-
微博賬號(hào)
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競(jìng)
物競(jìng)數(shù)據(jù)庫(kù)是一個(gè)全面、專業(yè)、專注,并且免費(fèi)的中文化學(xué)品信息庫(kù),為學(xué)生、學(xué)者、化學(xué)品研究機(jī)構(gòu)、檢測(cè)機(jī)構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺(tái)進(jìn)行交流。
數(shù)據(jù)庫(kù)采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國(guó)內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號(hào):物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)

-
微博賬號(hào):wjhxp
聯(lián)系我們
上海市延長(zhǎng)路149號(hào)上海大學(xué)科技園412室
公司總機(jī): 021-56389801
訂購(gòu)電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競(jìng)數(shù)據(jù)庫(kù) 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫(kù) | 關(guān)于物競(jìng) | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號(hào)
滬公網(wǎng)安備 31010602001115號(hào)