窄帶隙高分子光伏材料
- 2012-07-23
- 專題
由于能源緊張加上高分子光伏電池具有易加工、質(zhì)量小、成本低等優(yōu)點,各國越來越重視高分子太陽能電池的研究。高分子光電池之所以一直沒有大規(guī)模實際運用,是因為光電轉(zhuǎn)化效率還較低,迄今最高只有3.5%[1]。其主要原因是大多數(shù)活性材料最大吸收波段在350~650nm之間,而太陽光最大光子流在600~800nm,因此吸收光譜和太陽能不匹配;其次是光生載流子從活性材料傳輸?shù)诫姌O的損失,造成總能量效率一直不高。
研究窄帶隙的高分子光伏材料和能吸光的受體材料可以增加對太陽光的利用率,在活性材料中具有電子和空隙各自傳輸通道可以克服載流子傳輸損失。本文主要回顧了近年來出現(xiàn)的窄帶隙光伏材料,總結(jié)了物質(zhì)帶隙和分子鏈結(jié)構(gòu)的關(guān)系,提出合成窄帶隙光伏材料的一般思路;同時還對能夠吸光的受體材料和能夠形成載流子傳輸通道的“雙纜”光伏材料進行了介紹。
1 窄帶隙高分子光伏材料
盡管高分子光伏材料種類比較多,但由于性能和合成及加工方面的限制,實際應(yīng)用并不多,僅限于不多的幾種物質(zhì)及其衍生物。以PPV 衍生物研究得最多,其中聚3-己基噻吩(P3HT)、MEH-PPV[56]、OC1C10-PPV(MDMO-PPV)等效果最為突出。但它們最大吸收波長在 500nm,不太令人滿意。由于大多數(shù)活性材料最大吸收波段在350~650nm 之間,所以應(yīng)該研究最大吸收波長在紅光和近紅外的具有窄能隙(<1.8eV)的共軛高分子。
1.1 高分子光伏材料帶隙的影響因素
前人已經(jīng)進行大量研究的高分子帶隙 Eg和結(jié)構(gòu)的關(guān)系主要有以下幾點:(1)帶隙和共軛鏈的交替鏈長有關(guān),長度越短,帶隙越小;醌式結(jié)構(gòu)的分子鏈可以減少交替鏈長度,因此能夠減少共軛高分子的帶隙;(2)分子鏈中如果存在共軛的芳香單元,由于環(huán)間可能存在環(huán)扭轉(zhuǎn)張力,因此影響共軛,不利于帶隙的減少;(3)在有芳香單元的共軛鏈中,帶隙還和芳香性有關(guān)。芳香性體現(xiàn)了環(huán)中π電子定域和離域的競爭,對局部芳香體系來說,芳香性越強,電子在芳香體系內(nèi)離域越好。但對整個高分子分子鏈,如果芳香性太強,反而不利于π 電子在整個分子鏈中的離域;(4)帶隙和分子鏈間作用力有一定關(guān)系。分子間相互作用越強,帶隙越小。因此,通常物質(zhì)在固體下比在液態(tài)下有更低的帶隙,有序的相結(jié)構(gòu)比無序的相結(jié)構(gòu)有更低的帶隙;(5)如果共軛鏈中存在大體積的單元或取代基,因空間位阻的影響會降低共軛效果,使共軛長度減少,導(dǎo)致帶隙增加。總之,線性共軛高分子的帶隙(Eg)可用下式表[2]
Eg=E(r) + E(θ)+ E(Res) + E(Sub) + E(Int)
式中各項表示各種影響因素對帶隙的貢獻值。E(r)與交替出現(xiàn)的單雙鍵長度差有關(guān);E(θ)由空間位阻等原因使得芳香環(huán)共軛體系不能很好地共平面所產(chǎn)生的扭轉(zhuǎn)能決定;E(Res)是由芳香環(huán)的芳香共振能決定;E(Sub)代表了取代基對帶隙的貢獻;E(Int) 表示分子間相互作用的強弱對帶隙的影響。雖然高分子光伏材料和普通導(dǎo)電高分子有所不同,但在考慮帶隙和結(jié)構(gòu)的關(guān)系時,基本原則是一致的。
1.2 給吸電子單元交替對帶隙的影響
分子鏈由給電子單元和受電子單元交替構(gòu)成時,可以使它們之間的單鍵的電子發(fā)生偏移,使之具有部分雙鍵特征,從而減少交替鍵長,減小帶隙。常用的吸電子單元有如 2,1,3-苯并噻二唑(B)、噻吩并[3,4-b]吡嗪、苯并[1,2-c:4,5-c']雙[1,2,5]噻二唑、[1,2,5]噻二唑并[3,4,-g]喹喔啉;給電子單元有如雙噻吩、N,N-二甲基雙吡咯、2,5-二(2-噻吩基)- N -十二烷基吡咯(TPT)[3,4]。由于需要和受體混合,通常不能通過電化學(xué)聚合,因此尋找可溶的低帶隙的 D-A 高分子非常困難,僅有少數(shù)物質(zhì)[3]。
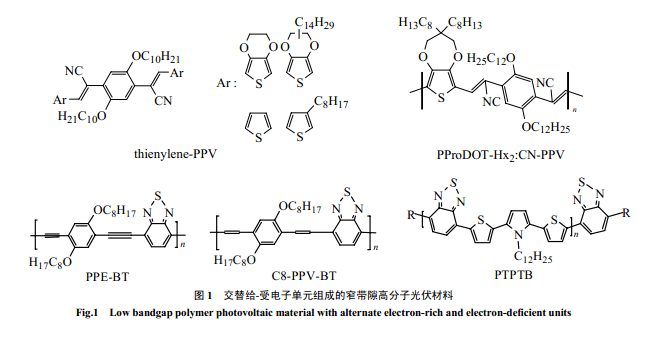
高分子能通過增加其最高占有軌道(HOMO)能量(減少氧化電位)或降低最低空軌道(LUMO)能量(減少還原電位)來減少帶隙Eg,但是,前者會降低開路電壓[3](分散異質(zhì)結(jié)光伏電池 VOC主要是由給體的HOMO和受體的LUMO 能量差決定的[5]),后者則會減少在異質(zhì)電池中激發(fā)態(tài)的電子轉(zhuǎn)移到受體的驅(qū)動力;同時,太低帶隙能夠增強輻射衰退,影響激發(fā)態(tài)壽命和激子擴散長度[3]。因此,高分子光伏材料不同于本征導(dǎo)電高分子,并非帶隙越低越好。Dhanabalan 等[3,6,16]考慮了這幾方面的因素,選用了中強的給電子單元 TPT 和受電子單元B 合成了D-A交替構(gòu)成的窄帶隙的PTPTB(1.6eV),吸收可以延到750nm,構(gòu)成的(PTPTB/PCBM)異質(zhì)電池能量轉(zhuǎn)換效率最高可以達到1%(AM=1.5)。路勝利等用給電子的 PPE單元[51]和PPV 單元[49]與2,1,3-苯并噻二唑共聚分別得低帶隙的交替共聚物PPE-BT和C8-PPV-BT(1.77eV),吸收都分別比純 PPE 和PPV 有較大的紅移。
也可以通過引入取代基來改變單元的親電性,在共軛鏈上分別交替引入給、受電子取代基同樣可以減少帶隙。2004年,Colladet等[7]合成了一系列 thienylene-PPV 衍生物窄帶隙光伏高分子(1.55-1.94eV),由于它在 PPV 的苯撐上交替引入了受電子的氰基和給電子的烷氧基,所合成的高分子的分子量高且有較好的溶解性。Barry等[58]合成了PProDOT-Hx2:CN-PPV 共聚物,不僅帶隙低(1.7eV),而且在空氣中有很好的穩(wěn)定性。
1.3 分子鏈上醌式結(jié)構(gòu)對帶隙的影響
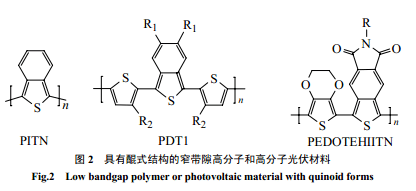
早在上世紀 80 年代中期,Wudl等[8,9]合成了窄帶隙的導(dǎo)電高分子聚苯并噻吩(PITN) ,噻吩環(huán)和苯環(huán)稠聯(lián)在一起,通過苯環(huán)來穩(wěn)定醌式結(jié)構(gòu),從而使主鏈中醌式結(jié)構(gòu)多于芳香結(jié)構(gòu)。由于醌式結(jié)構(gòu)有更低的帶隙,因此降低了 PITN的帶隙[10~12],雙甲基、雙甲氧基取代都不會影響此效果[12]。隨后一系列 PITN 衍生物,包括含有噻吩的窄帶隙材料相繼合成,但醌式結(jié)構(gòu)可使分子鏈具有剛性,即使引入側(cè)鏈也不易溶解,不利于加工和應(yīng)用[15],實際很少應(yīng)用于光伏材料。為了解決此問題,一些能溶解的 PITN預(yù)聚體的合成[14,17],給 PITN衍生物用于光伏材料帶來了希望。PDT衍生物聚(噻吩-苯并噻吩)和聚(1,3-雙噻吩-苯并噻吩)[13,14]用于光伏材料取得了較好效果。Polec 等[18,20]用新的途徑合成了被烷氧基等取代的單體雙硫雜苯酞,通過熱聚合合成了可溶的PITN衍生物聚(5,6-二硫雜辛基苯并噻吩),可以作為窄帶隙光伏材料(1.2eV)。Cravino等[19]研究了帶隙為1.1eV的PEDOTEHIITN(圖2),它在太陽光最強光子流700nm處有很強的吸收,同時顯示了優(yōu)良的穩(wěn)定性、溶解性和薄膜形成性能,是很有希望的光伏材料。
1.4 分子鏈間作用力對帶隙的影響
分子鏈聚集態(tài)結(jié)構(gòu)的有序化,可以增加分子鏈間的作用力,減少帶隙。新型窄帶隙光伏材料PEOPT可以通過一定的物理處理,使結(jié)晶度增加( 結(jié)構(gòu)的有序化增加),帶隙從2.1eV減少到1.75eV[22,23]。聚噻吩衍生物的局部規(guī)整減少了空間阻礙使噻吩環(huán)扭轉(zhuǎn)較少,同時增加了分子鏈之間的作用,從而減少帶[18]。苯環(huán)取代的分子鏈的聚集態(tài)結(jié)構(gòu)具有一定的有序性,分子鏈間作用力增大,可以形成部分結(jié)晶結(jié)構(gòu),并且結(jié)晶程度還和苯環(huán)取代基上的柔性鏈取代位置(決定伸展方向)有關(guān)[24]。
拉電子的噻吩并[3,4-c][1,2,5]噻二唑衍生物(TTD)和2,1,3-苯并噻二唑(BTZ)單元的 S…N 作用力使得聚合物聚集態(tài)結(jié)構(gòu)有序化,從而對降低帶隙有利[2],因此在給受單元交替的光伏材料中引入此結(jié)構(gòu)能進一步降低帶隙,如果解決了溶解性問題,就有望在光伏材料中得到應(yīng)用。

1.5 分子鏈中窄帶隙單元對帶隙的影響
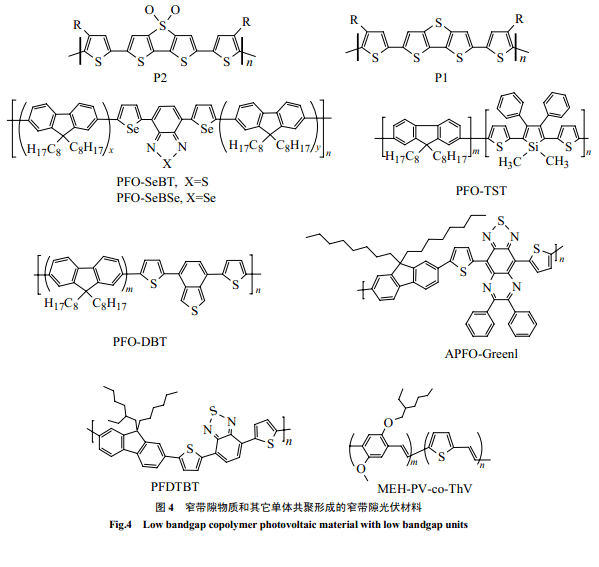
窄帶隙導(dǎo)電高分子通常不熔不溶,帶隙極窄,很難用于光伏材料。但可以通過和其它物質(zhì)共聚來改性,以滿足加工性、光電性等要求。聚芴是一種既有較高的空穴遷移率又比較穩(wěn)定的寬帶隙光電物質(zhì)。它可以通過和其它窄帶隙物質(zhì)共聚,進行性質(zhì)“剪裁”賦予共聚物以可能吸收紅光、吸收波段增寬和溶解性好等特點。窄帶隙物質(zhì)單元通常由給-受體單元交替構(gòu)成,噻吩由于具有較窄的帶隙和較高的穩(wěn)定性以及靈活的設(shè)計性能,一般作為給- 受體交替單元中的給電子體。Svensson 等[10]和周[25]分別合成了類似的新型光伏共聚物 PFDTBT 和PFO-DBT。它是由 9,9-二辛基芴和 7-雙-2-噻吩基-2,1,3-苯并噻二唑共聚而得,吸收延展到紅光,和PCBM 組成的異質(zhì)結(jié)電池轉(zhuǎn)換效率達到2%。芴與不同窄帶隙的給-受-給單元的共聚物 APFO-Green1[15,64,65]和APFO-Green2[67]都具有吸收紅移的特點,其中前者吸收更是達到 1000nm。Wan g等合成的PFO-TST[27]共聚物以及 PFO-SeBT(1.87eV)[26]和PFO-SeBSe[26,53](1.77eV) 共聚物,長波吸收增加,和 PCBM 組成的電池能量轉(zhuǎn)化效率分別達到了2.01% 和1%(AM=1.5)。
PPV 良好的光電性能和成膜性能一直倍受關(guān)注,Hou等[57]用帶隙較窄但成膜性能不好的聚 2,5-噻吩撐(PThV)和MEH-PPV共聚,得到的共聚物和 MEH-PPV比較,具有吸收紅移、吸收波段增寬以及空穴遷移率增大的優(yōu)點。Catellain等[28]通過共聚合成了P1、P2噻吩衍生共聚物,在窄帶隙物質(zhì)中引入烷基噻吩,使共聚物具有良好的溶解性,同時保持了窄帶隙,P2甚至達到了1.7eV。
1.6 共軛芳香環(huán)間雙鍵對帶隙的影響
高分子光伏材料研究最多、性能較好的是 PPV 衍生物:MEH-PPV、OC1C10-PPV(MDMO-PPV)等。其中(MDMO-PPV/PCBM)總能量轉(zhuǎn)化效率達到了迄今最高的 3.5%[1];這些物質(zhì)主鏈由芳香環(huán)和雙鍵交替構(gòu)成,形成所謂的苯撐結(jié)構(gòu)。由于存在雙鍵,減少了環(huán)的空間阻礙,抑制了環(huán)的扭曲,有利于共平面,使共軛長度增加,帶隙降低[11]。和PPV 相似,具有高分子量和高產(chǎn)量的聚噻吩撐(PTV)也具有此特點(1.55eV)[21]。現(xiàn)在設(shè)計低帶隙芳環(huán)共軛高分子光伏材料時,多引入雙鍵,以減少帶隙。
1.7 取代基對光伏材料帶隙的影響
共軛鏈上的取代基對光伏材料的性質(zhì)有很大的影響,它有以下幾個作用:(1)增加物質(zhì)的溶解性。如果光電材料不溶,難于合成和成型,應(yīng)用就有很大困難,沒有實際應(yīng)用價值。長鏈的線性烷基或烷氧基取代可以增加物質(zhì)的可溶性。(2)增加分子鏈的有序性。一些取代基如苯環(huán)取代能增加取代基間或鏈間相互作用,可以提高鏈結(jié)構(gòu)的規(guī)整性從而形成有序相,使帶隙減少。但是長的烷基取代不利于規(guī)整性,同時在合成時要避免相鄰取代基相撞,所謂形成頭碰頭(HH),尾碰尾(TT) 的缺陷從而造成空間位阻,影響分子鏈上鄰近芳香環(huán)的共平面性,減小有效共軛。Roncali等[29]通過對聚 3-烷基噻吩的研究,指出線性烷基 C 原子 7~9 個共軛效果最好,但支鏈取代效果不好,可能是由于不利于分子間相互作用。(3)給、受電子取代基可以分別使共軛系統(tǒng)的 HOMO,LUMO 升高和降低,降低帶隙。3,4- 乙烯基雙氧噻吩(EDT) 比噻吩給電子強,它的衍生物共聚合體有很窄的帶隙[31`33]和一定的溶解性[31],是很好的給電子體。
引入取代基不僅要考慮親電性和增溶性,還要考慮共軛效應(yīng)的影響[30]。側(cè)鏈芳香環(huán)可以和主鏈形成共軛,增加共軛效果,有利于減少帶隙。可用柔性烷基取代的苯基作主鏈的取代基,如PEOPT[22,23]。
當然,合成窄帶隙光伏材料時,不應(yīng)該考慮單一方面,各種影響因素都應(yīng)該考慮。
盡管隨著對窄帶隙高分子結(jié)構(gòu)的了解不斷加深,有一些窄帶隙的共軛高分子成功合成,但它們的外量子效率幾乎都不高,很少有超過50% 的,這仍然影響光電效率的提高。同時,由于受到不溶不熔及低分子量的困撓,實際上不能應(yīng)用于光伏材料[21]。因此合成具有良好溶解性,同時具有較高光電性能的高分子量的窄帶隙物質(zhì)仍然是一個挑戰(zhàn)性的工作。
2 具有吸光性的受電子物質(zhì)
受體物質(zhì)大多數(shù)對可見光的吸收很小,如聚對-吡啶乙烯撐(PpyV)[45]、具有n 型半導(dǎo)體性質(zhì)和低還原電位的含 N 雜環(huán)和主鏈有喹啉、嘧啶等結(jié)構(gòu)的共軛高分子[60];部分二酰亞胺(如PPEI[63])以及TiO2[62]等無機粒子雖可以作為受體材料,但對可見光吸收很低。由于受體是活性層的主體部分,如常用的C60 衍生物 C60 PCBM 占75% ,極大制約效率的提高。
Wienk等[59]用吸光性增強、對稱性不好的C70 PCBM 代替 C60 PCBM ,和 MDMO-PPV組成光電池,效率比 C60 PCBM 提高了50% 。高分子 BBL 能吸收紅光(740nm) ,和給體組成的活性材料有較強的光捕獲能力[46,47]。在共軛芳香環(huán)間的雙鍵上或苯環(huán)上引入拉電子氰基可以提高對電子的親和力,增加電子的遷移率,還可以提高陰離子的穩(wěn)定性,因此 CN-PPV[48]可作吸光的受體。Helmut 等[50]合成了活性物質(zhì) Pc-C60(最大吸收在兩個波段:<400nm,~700nm)可以作為分子給或受體光伏材料。但由于存在電荷傳輸障礙,構(gòu)造的光電池轉(zhuǎn)化效率并不好。此外,具有良好電子傳導(dǎo)性的無機納米半導(dǎo)體顆粒 CdSe也可以作為受體[61],它對長波可見光(720nm) 具有一定的吸收能力。

3 光伏材料中加入敏化劑增加光吸收
為了對太陽光進行“地毯”式的吸收,把窄帶隙和寬帶隙物質(zhì)混合構(gòu)成復(fù)合體,或把染料加入到共軛高分子中來增加對光的吸收。Winder[52]構(gòu)造了(PTPTB/PCBM+Nile 紅) 和(PTPTB/MDMO-PPV/PCBM)兩種結(jié)構(gòu)的光電池,發(fā)現(xiàn)前者效率有所提高,但后者卻有所下降。對于前者,PTPTB 和Nile 都吸紅光,貢獻了光電流,作用機理是否是染料把激發(fā)電子轉(zhuǎn)移給 PTPTB ,再通過 PTPTB 轉(zhuǎn)移給受體,還是兩者直接傳輸給 PCBM 還有待研究,但可以肯定染料形成的空穴的輸運有困難。對于后一種模型光電池,發(fā)現(xiàn)PTPTB 對光電流的貢獻遠不及 MDMO-PPV。可以看出,這種粗糙的混合,性能并沒有得到顯著改善。但可以預(yù)料,隨著對機理的不斷理解,在此基礎(chǔ)上進行改進,將是增加光利用率的一種有效途徑。
4 雙纜光伏材料
分子異質(zhì)高分子光伏電池是把結(jié)構(gòu)為給-受體的接枝或嵌段的共聚物作為光伏材料,通過一定的組裝技術(shù),給受體分別形成納米級各自連續(xù)兩相[34],構(gòu)成分別傳輸電子和空穴的兩種不同通道(圖6,7) ,即所謂的“雙纜”(雙通道)高分子異質(zhì)電池。通過精確控制給-受體嵌段長度,能夠使柱的直徑在激子的擴散距離內(nèi)。因為激子容易擴散到界面分離,電子、空穴有連續(xù)的通道傳輸?shù)礁髯噪姌O,所以有較高的轉(zhuǎn)換效率。目前已經(jīng)報道的大體有以下幾類。
(1)受體作為側(cè)鏈連在給體高分子骨架上。側(cè)鏈受體通常為C60 衍生物,主鏈為共軛的線性分子。2001 年Cravino等[35]把富勒烯吡咯烷通過用共價鍵和雙噻吩相連,合成了具有光電屬性的給體受體接枝單體。Ramosw 等[36]設(shè)計了(PPV 衍生物-C60)雙纜異質(zhì)光伏電池,獲得了 Voc 為830mV ,Isc 為420μA/cm2(AM1.5) ,F(xiàn)F為0.29 ,EQE 為6%(480nm) 。近年來,這種結(jié)構(gòu)的雙纜材料研究較多[37~39]。
(2)給體-受體都作為側(cè)鏈連在飽和脂肪鏈上。2004 年Lindner等[40]合成的嵌段共聚高分子具有骨架為飽和柔性σ 鏈,給體- 受體(雜環(huán)共軛體)以側(cè)鏈形式和主鏈相連,同時受體嵌段具有吸光的特點。
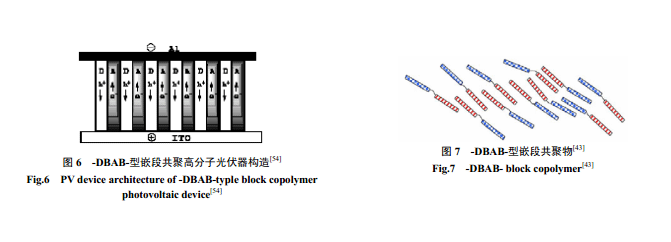
(3)-DBAB- 型線性嵌段共軛共聚物。2002 年Sun[43]設(shè)計了一種新的 DBAB 嵌段共軛共聚物(圖6、圖7) ,由于整個分子鏈都共軛,有利于電子傳輸。D是共軛給體嵌段,能吸收一定波長的光;A 是電子受體,也能吸收特定波長的光;B 是非共軛的絕緣柔性鏈(如脂肪單鏈),它能有效延緩電子、空穴的復(fù)合[44]。同時,σ鍵并不影響分子間和分子內(nèi)的電子轉(zhuǎn)移和激子的分離,這克服了-DA-型[41,42]電子和空穴復(fù)合太快太方便的缺點。此外,給-受體通過柔性鏈相連,還易于分子自組裝形成柱狀相分離。
5 高分子光伏材料發(fā)展方向和前景展望
為了提高太陽光的利用率和具有良好的載流子傳輸性能,高分子光伏材料可以從以下幾個方面進行深入研究:(1)研究量子效率高、加工性能好的窄帶隙光伏材料;(2)研究親電性強、電子遷移率高、吸光性強的高分子電子受體;(3)弄清楚染料敏化等具有多種波段吸光性的材料復(fù)合體的載流子傳輸機理和本質(zhì),以便采取措施減少載流子的陷阱;(4)深入研究分子結(jié)構(gòu)和聚集態(tài)結(jié)構(gòu)對吸光性、吸光波段的關(guān)系以及對載流子的傳輸?shù)挠绊懀瑥亩軌蚋鶕?jù)需要對光伏材料進行分子設(shè)計;(5)發(fā)展能夠精確控制形態(tài)的“雙纜”高分子光伏材料;(6)研究在氧氣和水的條件下具有一定穩(wěn)定性的光伏材料。
盡管高分子光伏材料已經(jīng)取得了不少進展,但仍然有一些問題需要克服。高分子光電材料雖然很多,但真正能運用到光伏電池上卻很有限,其中性能優(yōu)良的窄帶隙光伏材料更少,這極大制約了高分子光伏器件的發(fā)展和實際運用,因此開發(fā)新的高分子光伏材料特別是窄帶隙材料成為當務(wù)之急。同時,一些高分子光伏材料外量子效率不高,合成路線不穩(wěn)定可靠也不成熟,反應(yīng)活性不高,預(yù)聚體穩(wěn)定性或活性不高,合成的活性材料難溶難熔,不易加工,因此很難實際運用。 高分子光伏材料涉及到化學(xué)、光電子、固體物理等交叉學(xué)科,如果僅孤立從一個方面進行研究,是很難有突破的。但是,如今各種學(xué)科更多的研究者加入到該項研究中,對光伏材料的本質(zhì)有了越來越深入的理解,因此高分子光伏材料的突破性進展很有希望。
參考文獻
[1] F Padinger, R S Rittberger, N S Sariciftci. Adv. Funct. Mater., 2003, 13: 85~88.
[2] J Roncali. Chem. Rev., 1997, 97: 173~205.
[3] A Dhanabalan, J K J Van Duren, P A Van Hal et al. Adv. Funct. Mater., 2001, 11: 255~262.
[4] C Winder, N S Sariciftci. J. Mater. Chem., 2004, 14: 1077~1086.
[5] C J Brabec, A Cravino, D Meissner et al. Adv. Funct. Mater., 2001, 11: 374~380.
[6] J K J Van Duren, A Dhanabalan, P A Van Hal et al. Synth. Met., 2001, 121: 1587~1588.
[7] K Colladet, M Nicolas, L Goris et al. Thin Solid Films, 2004, 451-452: 7~11.
[8] M Kobayeshi, N Colaneri, M Boyse et al. J. Chem. Phys., 1985, 82: 5717~5723.
[9] J Wudl, M Kobayashi, A Heeger. Chem. Phys.,1984, 49: 3382~3384.
[10] J LJ bredas. Chem. Phys., 1985, 82: 3808~3811.
[11] C Quattrocchi, R Lazzaroni, J L Bredas et al. J. Phys. Chem., 1995, 99: 3932~3938.
[12] J L Bredas, A J Heeger , F Wudl. J. Chem. Phys., 1986, 85: 4673~4680.
[13] S E Shaheen, D Vangeneugden, R Kiebooms et al. Synth. Met., 2001, 121: 1583~1584.
[14] T S Hung, S A Chen. Polymer, 1999, 40: 3881~3884.
[15] X J Wang , E Perzon, J L Delgado et al. Appl. Phys. Lett., 2004, 85: 5081~5083.
[16] C J Braberc,C winder, N S Sariciftci et al. Adv. Funct. Mater., 2002, 12:709~712.
[17] H Meng, F Wudl. Macromolecules, 2001, 34: 1810~1816.
[18] I Polec, A Henckens,L Goris et al. J. Polym. Sci., Part A: Polym. Chem., 2003, 41: 1034~1045.
[19] A Cravino, M. A Loi, M C Scharber et al. Synth. Met., 2003, 137: 1435~1436.
[20] L Goris, M A Loi, H Neugebauer et al. Synth. Met., 2003, 138: 249~253.
[21] A Henckens, M Knipper, I Polec et al. Thin Solid Films, 2004, 451-452: 572~579.
[22] C J C Winder, M Scharber et al. Mater. Res. Symp. Proc., 2000, 598: BB3.24.1-BB3.24.6.
[23] C J Brabec, C Winder, M C Scharber et al. J. Chem. Phys., 2001, 115: 7235~7244.
[24] K E Aasmundtvei, E J Samuelsen, V V Mammo et al. Macromolecules, 2000, 33: 5481~5489.
[25] Q M Zhou, Q Hou, L P Zheng et al. Appl. Phys. Lett., 2004, 84: 1653~1655.
[26] R Q Yang, R Y Tian, J G Yan et al. Macromolecules, 2005, 38: 244~253.
[27] F Wang, J Luo, K Yang et al. Macromolecules, 2005, 38: 2253~2260.
[28] M Catellani, B Boselli, S Luzzati et al. Thin Solid Films, 2002, 403-404: 66~70.
[29] J Roncali, R Garreau, A Yassar et al. J. Phys. Chem., 1987, 91(27): 6706~6714.
[30] H A M Mullekom, J A J M Vekemans, E E E Havinga et al. Sci. Eng., 2001, 32: 1.
[31] L Groenendaal, F Jonas, D Freitag et al. Adv. Mater., 2000, 12: 481~494.
[32] S Akoudad, J Roncali. Synth. Met., 1999, 101: 149.
[33] D J Irvin, C J DuBois, J R Reynolds, Chem. Commun., 1999, 2121.
[34] B De Boer, U Stalmach, C Melzer et al . Synth. Met., 2001, 121:1541~1542.
[35] A Cravino, G Zerza, H Neugebauer et al. Synth. Met., 2001, 121: 1555~1556.
[36] A M Ramos, M T Rispens, J C Hummelen et al. Synth. Met., 2001, 119: 171~172.
[37] S Luzzati, M Scharber, M Catellani et al. Synth. Met., 2003, 139(3): 731~733.
[38] B De Boer, U Stalmach, C Melzer et al. Synth Met., 2001, 121: 1541~1542.
[39] V D V Marleen H, D B Bert, U Stalmach et al. Macromolecules, 2004, 37, 3673~3684.
[40] S M Lindner, M Thelakkat. Macromolecules, 2004, 37: 8832~8835.
[41] X L Chen ,S A Jenekhe. Macromolecules,1996, 29: 6189~6192.
[42] H Krueger, S Janietz, M Thesen et al. Proc. of SPIE, 2004, 5520: 98~109.
[43] S S Sun. Solar Energy Materials and Solar Cells, 2003, 79(2):257~264.
[44] S S Sun, Z Fan, Y Q Wang et al. Proc. SPIE, 2002, 4465: 121~128.
[45] K Tada, M Onoda, A A Zakhidov et al. Jpn. J. Appl. Phys., 1997,36: L306~L309.
[46] S A Jenekhe, S Yi. Appl. Phys. Lett., 2000 , 77(17) : 2635~2637.
[47] M M Alam, S A Jenekhe. Chem. Mater, 2004, 16(23): 4647~4656.
[48] M Granstr?m, K Petritsch, A C Arias et al. Synth. Met., 1999,102: 957~958.
[49] S G Lu, M J Yang, J Luo et al. Macromol. Chem. Phys., 2005, 206:664~671.
[50] H Neugebauer, M A Loi, C Winder et al. Sol. Energ. Mater. Sol. Cells., 2004, 83:201~209.
[51] S L Lu, M J Yang, J Luo et al. Synth. Met., 2004,140: 199~202.
[52] C Winder, G Matt, J C Hummelen et al. Thin Solid Films, 2002, 403-404: 373~379.
[53] 田仁玉,陽仁強,周清梅 等.華南理工大學(xué)學(xué)報(自然科學(xué)版), 2005, 33(2): 6~9.
[54] H Spanggaard, F C Krebs. Sol. Energ. Mater. Sol. Cells., 2004, 83: 125~146.
[55] M Al-Ibrahim, A Konkina, H K Roth et al. Thin Solid Films, 2005, 474: 201~ 210.
[56] 康博南,王立澤,邱勇. 科學(xué)通報, 2004, 49: 1611~1613.
[57] J H Hou, C H Yang, J Qiao et al. Synth. Met., 2005, 150:297~304.
[58] B C Thompson, Y G Kim, J R Reynolds et al. Macromolecules, 2005, 38: 5359~5362.
[59] M M Wienk, J M Kroon, W J H Verhees et al. Angew. Chem., Int. Ed, 2003, 42: 3371~3375.
[60] H Krueger, S Janietz, M Thesen et al. Proc. of SPIE, 2004, 5520: 98~109.
[61] B Sun, E Marx, N. C Greenham. Nano Lett., 2003, 3: 961~963.
[62] P Ravirajan, S A Haque, J R Durrant et al. J Appl Phys., 2004, 95: 1473~1480.
[63] J J Dittmer, K Petritsch, E A Marseglia et al. Synth. Met., 1999, 102: 879~880.
[64] M X Chen, E Perzon, N Robisson et al. Synth. Met., 2004, 146: 233~236.
[65] E Perzon, X J Wang, F L Zhang et al. Synth. Met., 2005, 154: 53~56.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
統(tǒng)計數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競化學(xué)品數(shù)據(jù)庫
-
物竟數(shù)據(jù)庫 手機版
國內(nèi)首款化工類專業(yè)手機應(yīng)用
-
微博賬號
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競
物競數(shù)據(jù)庫是一個全面、專業(yè)、專注,并且免費的中文化學(xué)品信息庫,為學(xué)生、學(xué)者、化學(xué)品研究機構(gòu)、檢測機構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺進行交流。
數(shù)據(jù)庫采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號:物競化學(xué)品數(shù)據(jù)庫

-
微博賬號:wjhxp
聯(lián)系我們
上海市延長路149號上海大學(xué)科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競數(shù)據(jù)庫 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫 | 關(guān)于物競 | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號
滬公網(wǎng)安備 31010602001115號