與胺/酰胺配體合作的雙功能分子催化
- 2013-01-21
- 專題
1. 介紹
過渡金屬類分子催化劑伴隨著新型的催化轉(zhuǎn)換以及人們對反應(yīng)機制的了解有了進一步的發(fā)展。特別是,持續(xù)付出的大量努力開發(fā)了具有兩個或者更多個活性位點協(xié)同一致的雙功能分子催化劑,從而實現(xiàn)了有機合成高效分子變換。最近,我們開發(fā)了金屬配體雙功能催化劑(協(xié)奏催化劑),其中non-innocent ligands直接參與基質(zhì)的激活和鍵的形成。雙功能分子催化是一個有吸引力和總體策略的概念可以實現(xiàn)有效地分子改造。1)
1995年, Noyori 和他的同事們報道了二胺配體在簡單的芳香酮和BINAP-Ru(II) [BINAP=2,2’-雙(二苯基膦基-1,1’-聯(lián)萘)]系統(tǒng)對映選擇性加氫方面一個嶄新的影響。2) RuCl2[(S)-binap](dmf)n,手性1,2-二胺和KOH組合在酮加氫反應(yīng)中比經(jīng)典的BINAP–Ru催化劑顯示出更為高級的活性。隨后,一個新的膦釕催化劑概念的雛形,以N-磺化1,2-二胺為手性試劑得到了進一步的發(fā)展,用于酮類的高效不對稱轉(zhuǎn)移氫化反應(yīng)。3)這個真正的催化劑的詳細的實驗內(nèi)容和理論分析表明,如Figure 1中所描述的,在循環(huán)催化過程中,具有正方形平面幾何狀的氨釕絡(luò)合物和配位飽和的氫化(胺)釕絡(luò)合物都有涉及。酰胺絡(luò)合物具有足夠的Br?nsted堿度,很容易氫化得到含有2-丙醇基的仲醇用以生產(chǎn)氫化(胺)配合物。NH單元被綁定到胺配合物金屬中心,具有足夠的酸性可以用來激活酮基質(zhì)。在氨基和胺絡(luò)合物的相互轉(zhuǎn)換中,金屬/NH部分協(xié)同合作通過一個環(huán)狀過渡態(tài)傳遞氫,其中,H–和H+ 被通過一個方式等量轉(zhuǎn)移,從氫(胺)絡(luò)合物到C=O鍵,或者從2-丙醇到胺絡(luò)合物,并且不用金屬中心的直接協(xié)調(diào)(外層機制)。雙功能過渡金屬分子催化劑這個獨特的概念使得反應(yīng)具有高利率和顯著的空間立體選擇性,因為 這個反應(yīng)是通過一個配置緊密的設(shè)備和手性催化劑得以完成。
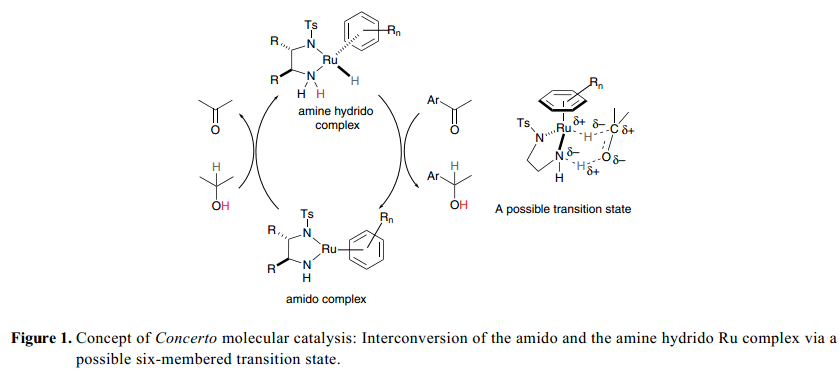
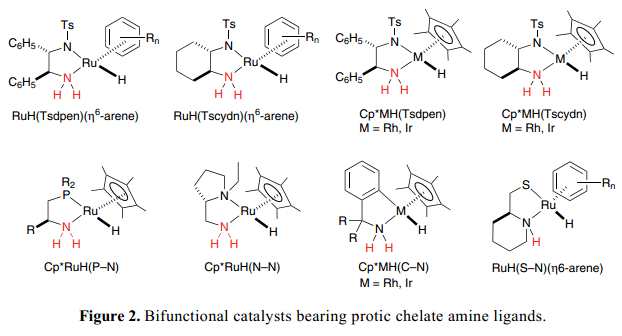
Figure 2 列舉了我們組新近開發(fā)的一些有明確定義的雙功能分子催化劑的代表性例子。手性雙功能η6-arene–Ru絡(luò)合物的概念是,RuH(Tsdpen)( η6-arene), [Tsdpen =N-(p-甲苯磺酰基)-1,2-二苯基乙二胺]已經(jīng)被成功的擴展到了類似的Cp*Rh和Ir絡(luò)合物中,Cp*MH(Tsdpen)[Cp*=η5-C(CH3)5,M=Rh, Ir]。4)除了N–磺酰化二胺復(fù)合物,8 和9組的金屬絡(luò)合物與二胺(N–N),5) 氨基膦(P–N),6) 氨基乙硫醇(S–N),7)o-氨基甲苯(C–N)8) 配體已經(jīng)被廣泛的開發(fā)用作完成金屬/NH效應(yīng)的雙功能分子催化劑。
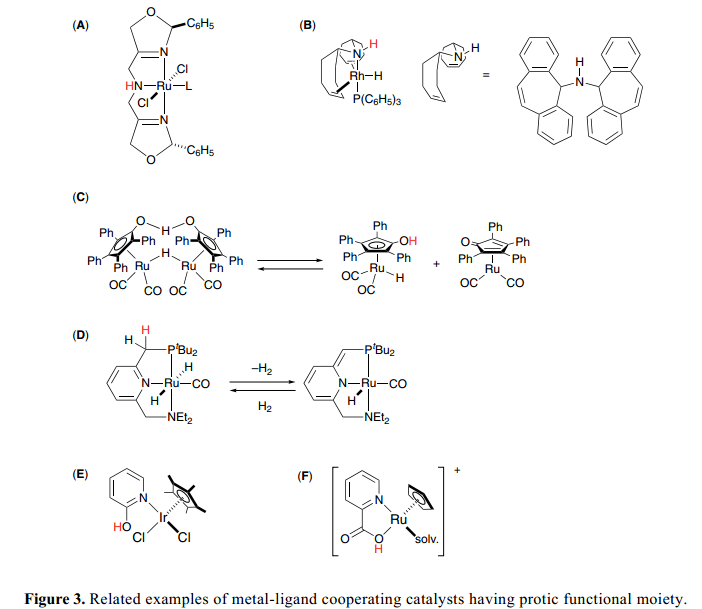
其他的研究小組研究別的相關(guān)的雙功能催化劑,Figure 3表示了幾組8 和9絡(luò)合物,其中輔助配體上的質(zhì)子官能團與金屬中心協(xié)作催化氫化,氫化轉(zhuǎn)移等。Zhang(A)9)和Grützmacher催化劑(B)10) 具有三叉質(zhì)子胺配體,能夠幫助H+/H–類似的方式下轉(zhuǎn)移到雙功能催化劑上面。Shvo催化劑 (C)11)可以釋放催化活性物質(zhì),環(huán)戊二烯和環(huán)戊二烯酮絡(luò)合物,OH基團作為質(zhì)子媒介在促進氫轉(zhuǎn)移過程中起到了一個關(guān)鍵的作用。最近,Milstein開發(fā)了新的PNN pincer complexes (D)12) 作為高活性氧化還原催化劑,并且找到了他們獨特的通過脫芳構(gòu)化作用的氫化/脫氫反應(yīng)機制。Fujita和Yamaguchi設(shè)計了Cp*Ir 絡(luò)合物(E)13),具有2-羥基吡啶和pyridonate配體,可以有效地通過酸性O(shè)H 官能團促進仲醇的脫氫反應(yīng)。Kitamura和Tanaka 證明了The acidic nature of CpRu fragment (F)上的2-喹啉羧酸酸性質(zhì)影響到了烯丙基醇C–O鍵的活性從而得到烯丙基醚。14)
本為關(guān)注于我們最新的進展,基于金屬/NH協(xié)作的雙功能分子催化劑,它們利用不對稱還原轉(zhuǎn)換反應(yīng)和相關(guān)的對映選擇性C–C和C–N鍵的生成反應(yīng)。
2.使用雙功能催化劑的不對稱轉(zhuǎn)移氫化反應(yīng)1c)
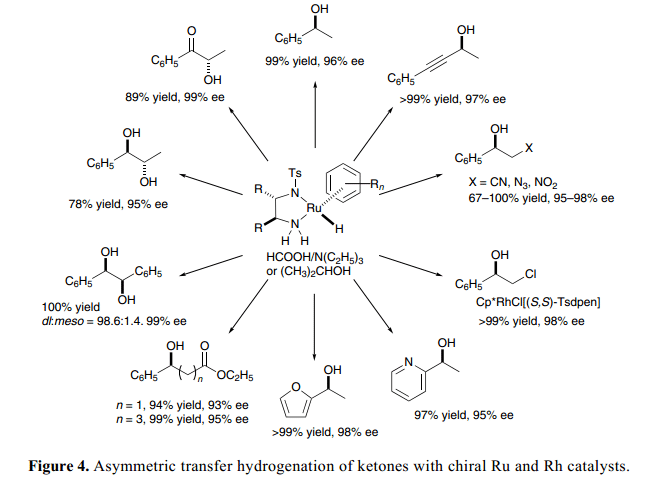
一般來講,2-丙醇可以用作一種安全、無毒、環(huán)境友好型氫源。盡管2-丙醇的不對稱還原得到了一個令人滿意的結(jié)果,但是,其內(nèi)在的一個弊端是氫轉(zhuǎn)移是可逆的,這就導(dǎo)致了轉(zhuǎn)移的局限取決于系統(tǒng)的熱力學(xué)因素以及反應(yīng)混合物和催化劑在長時間曝光下產(chǎn)品對映體純度的損耗。另一方面,甲酸遵循不對稱還原反應(yīng),在不可逆轉(zhuǎn)方法中轉(zhuǎn)換率100%。15)
簡單的芳香酮類和含有雙功能催化劑的甲酸、三乙胺混合物的不對稱還原反應(yīng)具有高效活性、選擇性、廣泛的底物范圍以及實用性等特點。雙功能氫化(胺)-Ru配合物是配體飽和,催化系統(tǒng)容許有氨基、酯、氰基、硝基、疊氮化和氯基團以及芳香雜環(huán)和炔鏈,1c)如Figure 4總結(jié)所示。C–N雙鍵也可以與甲酸和三乙胺含有手性Ru 或者Rh 催化劑的混合物還原得到相應(yīng)的胺,具有一個很高的對映體過量百分數(shù)。16)
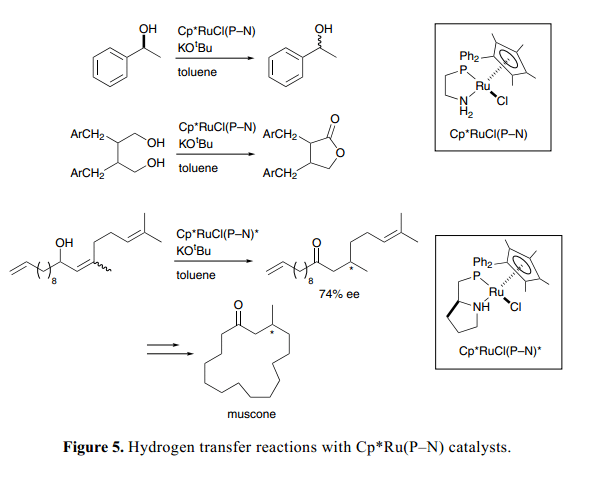
如Figure 5所示,一系列的含有質(zhì)子膦胺螯合物配體的Cp*Ru(P–N)絡(luò)合物 也用作氫化轉(zhuǎn)移的高效催化劑。基于可逆氫化機制通過胺和氨絡(luò)合物兩者之間互換,氧化還原過程可以成功的用于手性醇的快速外消旋化17)以及利用丙酮將二醇特定的選擇氧化為內(nèi)酯。18)手性Cp*Ru(P–N)復(fù)雜絡(luò)合物對映選擇性異構(gòu)化烯丙基醇為β-取代基酮類,接下來閉環(huán)置換得到溫和的(R )-麝香酮。19)
3. 極性基質(zhì)雙功能催化劑的催化加氫6)
由于我們在2001年報道了醇溶劑在綁定異裂H2鍵到16-電子[Cp*Ru{Me2 N(CH2)2 NH2}]+(Cp*Ru(N–N))片段的的影響,20)我們系統(tǒng)的測試了除酮以外的極性基質(zhì)的催化氫化,發(fā)現(xiàn)具有叔膦基替換叔胺可以導(dǎo)致Ru/NH 雙功能范圍擴大化。例如, Cp*Ru(P–N復(fù)合物,[Cp*Ru{Ph2 P(CH2)2 NH2}]+, 在各種終端環(huán)氧化物中給不飽和C–O選擇性提供氫鍵,從而生成相應(yīng)的仲醇,然而Cp*Ru(N–N) 復(fù)合物是完全消極惰性的。21)
Cp*RuCl(P–N)催化劑直接催化氫化N-acylcarbamates和carboxyimides選擇性得到N -protected amines 和alcohols。22)
值得注意的是,目前的氫化作用適用于還原轉(zhuǎn)化反應(yīng)。雙功能催化條件下伯胺從N-phthalimides脫保護同樣適用于Gabriel氨基酸的合成。例如, N-鄰苯二甲酰-L-苯丙氨酸甲酯通過手性N–acyloxazolidiones處理氫解得到物光學(xué)純度損耗的L-Phe甲酯鹽酸鹽,這是由Evans 研發(fā)的合成手性醇和無光學(xué)純度損耗的original 手性助劑的非常有用的合成中間體,如Figure 6所示。23a)
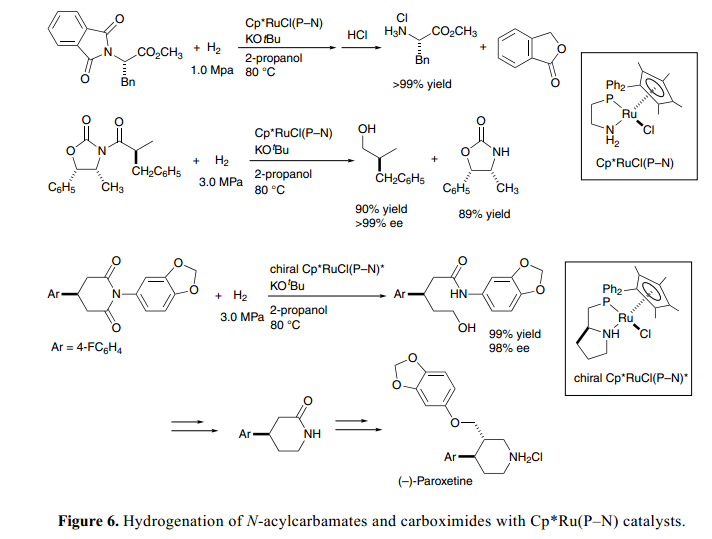
手性Cp*Ru(P–N)催化劑,具有手性P–N配合基鍵(由L-普氨酸衍生得到)促進前手性4-arylglutarimides對映選擇性氫化通過去保護得到相對應(yīng)的δ-羥基酰胺,具有很好的ee值,且產(chǎn)率很高。進一步細化加工得到手性哌啶酮衍生物,這個可以作用合成生理學(xué)光學(xué)活性手性化合物包括抗抑郁藥物帕羅西汀等的重要合成中間體。
Cp*Ru(P–N)催化劑強壓條件下也能促進酯和酰胺類氫化得到醇或者胺。23b)這個氫化過程使用金屬氫化試劑化學(xué)計量法也將為傳統(tǒng)的還原方法提供一個很強大的替代方法。
4. 雙功能催化劑有氧氧化反應(yīng)24)
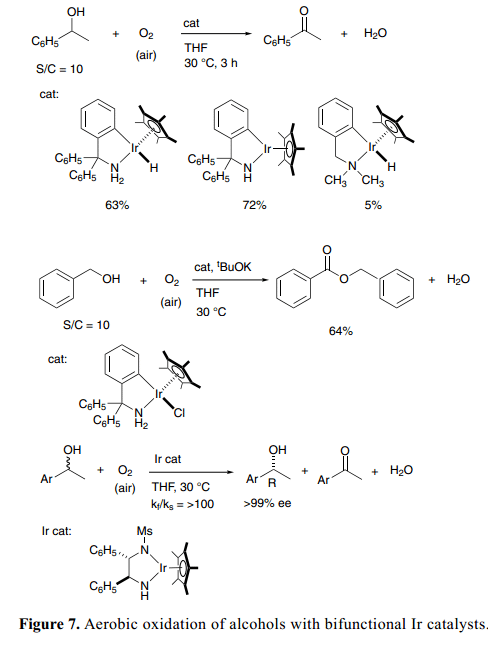
當分子氧用于雙官能團醇類脫氫氧化反應(yīng)中的氫原子受體時,有氧氧化變?yōu)橐粋€簡單且有機廢物最小化的一個過程。幸運的是,我們發(fā)現(xiàn)了一個新的可孤立的官能團酰胺-Ir絡(luò)合物,具有一個C–N螯合胺配體Cp*Ir[ κ2(N,C)-(NHCR2-2-C6H4)] (R=C6H5和CH3),和相應(yīng)的氫(胺)絡(luò)合物Cp*IrH[κ2(N,C)-{NH2CR2-2-C6H4}],可以作為酮類轉(zhuǎn)移氫化反應(yīng)的高效催化劑,促進催化醇類的有氧氧化。8a)1-苯基乙醇的催化反應(yīng)在空氣大氣壓強下平穩(wěn)進行,含有酰胺-Ir絡(luò)合物,Cp*Ir[κ2(N, C)-{NHC(C6H5)2-2-C6H4}] (Figure 7)。25)氫(胺)-Ir 絡(luò)合物,Cp*IrH[κ2(N,C)-{NH2C(C6H5)2-2-C6H4}], 和二元態(tài)催化劑系統(tǒng)(含有氯(胺)-Rh、-Ru絡(luò)合物和KOC(CH3)3), 也能得到氧化產(chǎn)物苯乙酮。伯醇在相同條件下反應(yīng)得到二聚產(chǎn)品、酯類等。當含有氯絡(luò)合物組分催化劑的芐醇混合物Cp*IrCl[κ2(N,C)-{NH2C(C6H5)2-2-C6H4}], 與當量的KOC(CH3)3在THF中攪拌反應(yīng),空氣溫度為30 °C時,得到相對應(yīng)的苯甲酸芐酯衍生物。
當應(yīng)用到二級醇與手性酰胺催化劑的動力學(xué)拆分時,醇的有氧氧化更為引起人們的興趣(Figure 7)。有氧氧化條件下通過手性Ir 絡(luò)合物Cp*Ir[( S,S)-Msdpen](Ms=甲磺酰),有效的解決了外消旋1-苯乙醇的動力學(xué)拆分,得到48%的 (R)-1-苯基乙醇和98% ee;kf/ ks比率為100。26)同樣的,1-indanol與1-tetralol 室溫下反應(yīng)得到R-enantiomers,>99% ee且46–50%產(chǎn)率。
5.承接金屬-N鍵的雙核雙功能催化劑的擴展27)
金屬配體雙能能效應(yīng)實現(xiàn)了單核half-sandwich氨基絡(luò)合物的形成,有望于 有效的制備具有M–N鍵的亞氨基-橋雙核絡(luò)合物。細致的結(jié)構(gòu)分析和NMR研究 表明了吸電子雙核集團使得亞氨基橋雙核絡(luò)合物穩(wěn)固,28) 同時也具有一源于M–N鍵的酸堿雙功能性質(zhì)。
不飽和雙(亞氨)-橋二銠(III)復(fù)合物配體,[(Cp*Rh)2(μ-NTs)2](Ts=SO2C6H4CH3-p)室溫下與H2(1atm)在CH2Cl2中反應(yīng)24h得到雙(亞氨)-橋二銠(II)復(fù)合物(Cp*Rh)2(μ-NHTs)2],產(chǎn)率為82% 如Figure 8所示。應(yīng)該注意的是,在這個反應(yīng)中, H2形式上轉(zhuǎn)換為兩個氨基質(zhì)子和兩個電子用于二銠在[(Cp*Rh)2(μ-NTs)2]的還原。雙(亞氨) 復(fù)合物與過量的2-丙醇反應(yīng)得到雙(胺基)復(fù)合物。兩個NH質(zhì)子的產(chǎn)生與金屬中心的兩個電子還原相結(jié)合與伯醇、仲醇通過不飽和單核氨基絡(luò)合物脫氫作用形成了鮮明的對比,這就導(dǎo)致了hydrido–amine復(fù)合物的形成,并且如上所述沒有形成正式的金屬的還原。
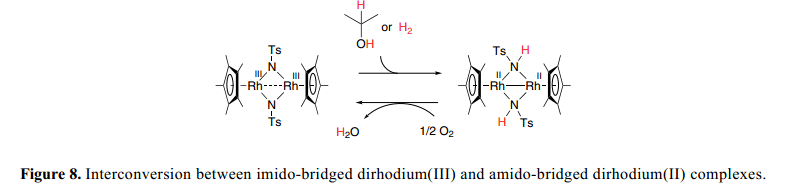
雙(氨基)-橋二銠(II)復(fù)合物與O2快速反應(yīng)形成雙(亞氨)-橋二銠(III)復(fù)合物和水。29) 此外,雙核imido和amido之間的溫和的氧化還原互換可以應(yīng)用到H2和醇的有氧氧化中起到催化作用。
6. 不對稱共軛加成雙功能催化劑30)
堿性氨基復(fù)合物可以與含酸性的有機化合物反應(yīng),得到含有金屬碳鍵親核試劑的胺復(fù)合物。例如,我們發(fā)現(xiàn)紫色Ru復(fù)合物,Ru[(R, R)-Tsdpen]( η6-三甲苯),–30°C條件下與丙二酸二甲酯在甲苯中平穩(wěn)反應(yīng)得到黃色結(jié)晶物,Ru[CH(CO2 CH3)2][(R,R)-Tsdpen](η6-三甲苯)。31)如果從一個氨基金屬復(fù)合物中得到的具有金屬鍵親核試劑的胺胺復(fù)合物進攻缺電子受體,催化作用的C–C 鍵形成得以實現(xiàn)。事實上,手性Ru催化劑,Ru[(S,S)-Tsdpen](η6-arene)有效的提高了dimethyl malonate對環(huán)enones 或者nitro olefins的對映選擇性Michael 加成反應(yīng)得到具有很好的ee值的加成產(chǎn)物(Figure 9)。通過雙功能Ru催化劑,1,3-dicarbonyl compounds對cyclic enones的Michael addition 反應(yīng)的NMR和DFT分析結(jié)果表明對映選擇性C–C鍵的形成通過螯合電子對中間物的形成, Ru金屬中心與enone分子協(xié)同為C–C鍵的形成制造出一個高組織性環(huán)境,只產(chǎn)生一種對映選擇性產(chǎn)品。31b)

手性氨基催化劑對映選擇性共軛加成可以擴展到α –cyanoacetates與dialkyl azodicarboxylates的Ir-催化反應(yīng)得到直接胺化產(chǎn)物。2)當乙炔酯用作親電子試劑,α–cyanoacetate和雙功能手性催化劑對映選擇性和Z 選擇性共軛加成得到手性加成產(chǎn)物,擁有一個四元碳中心,具有很好的ee值。33)
7. 總結(jié)
本文主要關(guān)注最新進展的化學(xué)概念上的新的包括立體性、區(qū)域性和化學(xué)選擇性還原劑或者氧化轉(zhuǎn)移以及對映選擇性質(zhì)C–C、C–N鍵(通過Michael-type共軛加成而產(chǎn)生)的協(xié)作催化劑與雙功能過渡金屬分子催化劑。配位胺試劑合理的設(shè)計使得M/NH單元中電子因素在雙功能催化劑中顯示平衡狀態(tài),是其具有廣泛性和高實用性的催化性能的決定性因素。我們相信目前雙功能分子催化劑能夠為開啟新的選擇性分子轉(zhuǎn)移包括對映選擇性合成的層面上提供一個廣闊的機會。
參考文獻
1) a) T. Ikariya, K. Murata, R. Noyori, Org. Biomol. Chem. 2006, 4, 393. b) K. Mu?iz, Angew. Chem. Int. Ed. 2005, 44 , 6622. c) T. Ikariya, A. J. Blacker, Acc. Chem. Res. 2007, 40, 1300. d) T. Ikariya, Bull. Chem. Soc. Jpn. 2011, 84 , 1.
2) a) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1995, 117, 2675. b) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1995, 117, 10417.
3) a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1995 , 117 , 7562. b) K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya, R. Noyori, Angew. Chem. Int. Ed. Engl. 1997, 36 , 285.
4)a) K. Murata, T. Ikariya, R. Noyori, J. Org. Chem. 1999, 64, 2186. b) K. Mashima, T. Abe, K. Tani, Chem. Lett. 1998, 1199. c) K. Mashima, T. Abe, K. Tani, Chem. Lett. 1998, 1201.
5) M. Ito, M.Hirakawa, K. Murata, T. Ikariya , Organometallics 2001, 20 , 379.
6) a) M. Ito, T. Ikariya, Chem. Commun. 2007, 5134. b) M. Ito, A. Osaku, C. Kobayashi, A. Shiibashi, T. Ikariya, Organometallics 2009, 28 , 390. c) M. Ito, T. Ikariya, J. Synth. Org. Chem. Jpn. 2008, 66 , 1042.
7) a) M. Ito, Y. Shibata, A. Watanabe, T. Ikariya, Synlett 2009, 1621. b) M. Ito, A. Watanabe, Y. Shibata, T. Ikariya, Synlett 2010, 29 , 4584.
8) a ) S. Arita, T. Koike, Y. Kayaki, T. Ikariya , Organometallics 2008, 27 , 2795. b) J.-B. Sortais, V. Ritleng, A. Voelklin, A. Holuigue, H. Smail, L. Barloy, C. Sirlin, G. K. M. Verzijl, J. A. F. Boogers, A. H. M. de Vries, J. G. de Vries, M. Pfeffer, Org. Lett. 2005, 7, 1247.
9) Y. Jiang, Q. Jiang, X. Zhang, J. Am. Chem. Soc. 1998, 120, 3817.
10) a) P. Maire, T. Büttner, F. Breher, P. Le Floch, H. Grützmacher, Angew. Chem. Int. Ed. 2005, 44 , 6318. b) T. Zweifel, J.-V. Naubron, T. Büttner, T. Ott, H. Grützmacher, Angew. Chem. Int. Ed. 2008, 47 , 3245. c) T. Zweifel, J.-V. Naubron, H. Grützmacher, Angew. Chem. Int. Ed. 2009 , 48 ,
559.
11) a ) Y . B l u m , D . C z a r k i e , Y . R a h a m i n , T . S h v o , Organometallics 1985, 4, 1459. b) Y. Shvo, D. Czarkie, Y. Rahamim, D. F. Chodosh, J. Am. Chem. Soc. 1986, 108, 7400.
12) a) J. Zhang, G. Leitus, Y. Ben-David, D. Milstein, J. Am. Chem. Soc. 2005, 127, 10840. b) C. Gunanathan, Y. Ben-David, D. Milstein, Science 2007, 317, 790. c) S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron, D. Milstein, Science 2009, 324, 74.
13) a) K. Fujita, N. Tanino, R. Yamaguchi, Org. Lett. 2007, 9, 109. b) R. Yamaguchi, C. Ikeda, Y. Takahashi, K. Fujita, J. Am. Chem. Soc. 2009, 131, 8410.
14) a) S. Tanaka, H. Saburi, Y. Ishibashi, M. Kitamura, Org. Lett. 2004, 6, 1873. b) S. Tanaka, H. Saburi, M. Kitamura, Angew. Chem. Int. Ed. 2005, 44 , 1730. c) S. Tanaka, T. Seki, M. Kitamura, Angew. Chem. Int. Ed. 2009, 48 , 8948.
15) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1996, 118, 2521.
16) a) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1996 , 118 , 4916. b) J. Mao, D. C. Baker, Org. Lett. 1999, 1, 841.
17) M. Ito, A. Osaku, S. Kitahara, M. Hirakawa, T. Ikariya, Tetrahedron Lett. 2003, 44 , 7521.
18) a) M. Ito, A. Osaku, A. Shiibashi, T. Ikariya, Org. Lett. 2007, 9, 1821. b) M. Ito, A. Shiibashi, T. Ikariya, Chem. Commun. 2011, 2134.
19) M. Ito, S. Kitahara, T. Ikariya, J. Am. Chem. Soc. 2005, 127, 6172.
20) M. Ito, M. Hirakawa, K. Murata, T. Ikariya , Organometallics 2001, 20 , 379.
21) M. Ito, M. Hirakawa, A. Osaku, T. Ikariya, Organometallics 2003, 22 , 4190.
22) a) M. Ito, A. Sakaguchi, C. Kobayashi, T. Ikariya, J. Am. Chem. Soc. 2007, 129, 290. b) M. Ito, C. Kobayashi, A. Himizu, T. Ikariya, J. Am. Chem. Soc. 2010, 132, 11414.
23) a) M. Ito, L. W. Koo, A. Himizu, C. Kobayashi, A. Sakaguchi, T. Ikariya, Angew. Chem. Int. Ed. 2009, 48 , 1324. b) M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu, T. Ikariya, J. Am. Chem. Soc. 2011, 133, 4240.
24) T. Ikariya, S. Kuwata, Y. Kayaki, Pure Appl. Chem. 2010, 82, 1471.
25) S. Arita, T. Koike, Y. Kayaki, T. Ikariya, Chem. Asian J. 2008, 3, 1479.
26) S. Arita, T. Koike, Y. Kayaki, T. Ikariya, Angew. Chem. Int. Ed. 2008, 47 , 2447.
27) S. Kuwata, T. Ikariya, Dalton Trans. 2010, 39 , 2984.
28) a) K. Ishiwata, S. Kuwata, T. Ikariya, Organometallics 2006, 25 , 5847. b) H. Arita, K. Ishiwata, S. Kuwata, T. Ikariya, Organometallics 2008, 27 , 493.
29) K. Ishiwata, S. Kuwata, T. Ikariya, J. Am. Chem. Soc. 2009 , 113, 5001.
30) a) T. Ikariya, I. D. Gridnev, Chem. Rec. 2009, 9, 106. b) T. Ikariya, I. D. Gridnev, Top. Catal. 2010, 53 , 894.
31) a) M. Watanabe, K. Murata, T. Ikariya, J. Am. Chem. Soc. 2003, 125, 7508. b) I. D. Gridnev, M. Watanabe, H. Wang, T. Ikariya, J. Am. Chem. Soc. 2010, 132, 16637.
32) Y. Hasegawa, M. Watanabe, I. D. Gridnev, T. Ikariya, J. Am. Chem. Soc. 2008, 130, 2158.
33) Y. Hasegawa, I. D. Gridnev, T. Ikariya, Angew. Chem. Int. Ed. 2010, 49 , 8157.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
統(tǒng)計數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競化學(xué)品數(shù)據(jù)庫
-
物竟數(shù)據(jù)庫 手機版
國內(nèi)首款化工類專業(yè)手機應(yīng)用
-
微博賬號
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競
物競數(shù)據(jù)庫是一個全面、專業(yè)、專注,并且免費的中文化學(xué)品信息庫,為學(xué)生、學(xué)者、化學(xué)品研究機構(gòu)、檢測機構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺進行交流。
數(shù)據(jù)庫采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號:物競化學(xué)品數(shù)據(jù)庫

-
微博賬號:wjhxp
聯(lián)系我們
上海市延長路149號上海大學(xué)科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競數(shù)據(jù)庫 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫 | 關(guān)于物競 | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號
滬公網(wǎng)安備 31010602001115號