配位吸附
- 2012-12-11
- 專題
配位吸附(也稱配體吸附) 是將具有配位性能的金屬離子載于樹脂上,通過(guò)金屬離子與吸附質(zhì)(配體性質(zhì)的化合物) 間的配位作用而將吸附質(zhì)吸附在樹脂上。具有配位吸附功能的樹脂稱為配位吸附樹脂。由于配位吸附大多在水介質(zhì)中進(jìn)行,而水是一種弱的配體,會(huì)與樹脂上的金屬離子發(fā)生較弱的配位作用。當(dāng)樹脂在吸附其它配體時(shí),水從金屬離子上被置換下來(lái),因此,配位吸附又稱為配體交換吸附[1,2],配位吸附樹脂也被稱為配體交換樹脂。配位吸附的研究已經(jīng)歷了半個(gè)世紀(jì)的歷程,現(xiàn)將配位吸附的研究作全面的介紹。
1 配位吸附發(fā)展簡(jiǎn)史
1952年,美國(guó)的Walton和他的學(xué)生 Stokes,將Cu2+和Ag+分別載于磺酸型和丙烯酸型離子交換樹脂上,并研究了負(fù)載Cu2+和Ag+之后的樹脂對(duì)水中的氨、正丁胺、 哌啶、苯胺和乙二胺的吸附情況。1953年,他們?cè)诿绹?guó)化學(xué)學(xué)會(huì)第123 屆會(huì)議上公布了部分結(jié)果,并于次年將完整的結(jié)果公開發(fā)表[3]。但他們的這一研究結(jié)果在以后的近 10年時(shí)間里并未引起多大的反響。1961年美國(guó)的Helfferich發(fā)現(xiàn)了配體交換樹脂在色譜分離上的重要應(yīng)用,并在《Nature 》上發(fā)表了關(guān)于配體交換色譜技術(shù)的文章[1]。接著他研究了配體吸附和配體交換的平衡,從而奠定了配位吸附的熱力學(xué)基礎(chǔ)[4]。隨后大量的關(guān)于配體交換色譜的研究結(jié)果發(fā)表在《J. Chromatography 》等雜志上。被分離的物質(zhì)包括氨基酸[5]、核酸和核苷酸[6~8]、生物堿[9,10]、芳胺[11~15]、脂肪酸[16]、醇和糖[17]、脂肪胺[18,19]和硫化物[20]等。上個(gè)世紀(jì)六七十年代是配體交換色譜研究最活躍的時(shí)期,70年代末期這方面的論文數(shù)量開始減少。就在配體交換色譜的研究高潮尚未過(guò)去的時(shí)候,70年代初期,另一位離子交換與吸附樹脂研究領(lǐng)域的杰出人物,后交聯(lián)技術(shù)的發(fā)明者,前蘇聯(lián)的Davankov開創(chuàng)了將配體交換色譜應(yīng)用于手性氨基酸異構(gòu)體拆分的應(yīng)用領(lǐng)域。他將手性氨基酸的一個(gè)異構(gòu)體,如L-脯氨酸,接到氯甲基化的聚苯乙烯樹脂上,再通過(guò)L-脯氨酸與Cu2+間的配位作用而將Cu2+載于樹脂上。當(dāng)含有DL-脯氨酸外消旋體的溶液流過(guò)此樹脂的吸附柱時(shí),D-脯氨酸被優(yōu)先吸附在柱上,而L- 脯氨酸的吸附很弱,能直接流過(guò)色譜柱,或用較低濃度的氨水即被洗脫下來(lái)。D-脯氨酸的洗脫需要用濃度較高的氨水[21,22]。采用這種手性配體交換樹脂很容易將手性氨基酸對(duì)映體分開。將一系列手性氨基酸或非氨基酸手性物質(zhì)載于樹脂上,再負(fù)載不同的金屬離子,可以實(shí)現(xiàn)對(duì)多種手性氨基酸的拆分[23~27]。Davankov開創(chuàng)了這一領(lǐng)域之后,大量學(xué)者開始從事這方面的研究工作。經(jīng)過(guò)約10年左右的時(shí)間,到上世紀(jì)80年代后期,手性配體交換樹脂對(duì)手性氨基酸拆分的研究報(bào)道逐漸減少。
上世紀(jì)90年代初到現(xiàn)在的10年時(shí)間里,配位吸附方面的文獻(xiàn)報(bào)道主要集中在如下幾個(gè)方面:(1)采用配位吸附來(lái)富集和回收廢水和海水中微量的砷化物[28~31];(2)配位吸附和電荷作用相結(jié)合,去除和回收水溶液中微量的負(fù)離子[33~36];(3) 采用配位吸附樹脂分離胺類化合物[14,15]。
2 配位吸附的優(yōu)缺點(diǎn)
2.1 配位吸附的優(yōu)點(diǎn)
2.1.1較高的吸附強(qiáng)度 由于配位鍵的強(qiáng)度較高,配位吸附也可以達(dá)到較高的吸附強(qiáng)度[1,2]。
2.1.2 較好的吸附選擇性 配位吸附樹脂只能吸附含配位原子的化合物。因此,它可以很容易地從不含配位原子的化合物中分離出具配位能力的化合物。另外對(duì)于含配位原子的化合物,由于其本身配位能力的差異,或配位時(shí)空間位阻的不同,配位吸附的選擇性也出現(xiàn)很大的差別。因而采用配位吸附有時(shí)很容易將許多結(jié)構(gòu)和性質(zhì)較相近的化合物分離開來(lái)。
2.1.3 容易洗脫 一般說(shuō)來(lái),氨水可將絕大多數(shù)被吸附的配體很容易地洗脫下來(lái),這是由于氨本身是一種強(qiáng)的配體,容易將吸附質(zhì)置換下來(lái)。由于氨極易揮發(fā),一般情況下被洗脫下來(lái)的吸附質(zhì)經(jīng)過(guò)干燥即可除去其中的氨[4]。不同的配體,由于它們?cè)诮Y(jié)構(gòu)上的差異,被吸附的強(qiáng)度也有一定的差異,調(diào)節(jié)氨水的濃度可將吸附強(qiáng)度不同的各種吸附質(zhì)依次洗脫下來(lái)。
2.2 配位吸附的缺陷
2.2.1 金屬離子的脫落 在水介質(zhì)中,被負(fù)載的金屬離子會(huì)不同程度地從樹脂上脫落下來(lái)進(jìn)入吸附介質(zhì)或洗脫液中,脫落的原因是水中不可避免地存在一些正負(fù)離子,其中的陽(yáng)離子會(huì)與樹脂上負(fù)載的金屬離子發(fā)生交換,從而使被負(fù)載的金屬離子從樹脂上脫落下來(lái)。采用對(duì)金屬離子結(jié)合能力較強(qiáng)的樹脂,如螯合樹脂作配位吸附樹脂的基體,可以減少金屬離子脫落的可能。金屬離子的脫落會(huì)帶來(lái)不利的后果:(1)會(huì)使被洗脫的物質(zhì)受到金屬離子的污染,需要進(jìn)一步處理,以去除這些金屬離子;(2)金屬離子的脫落會(huì)使樹脂上負(fù)載的離子不斷減少,使用的過(guò)程中或使用一段時(shí)間之后,樹脂上的金屬離子須不斷地補(bǔ)充,這會(huì)給實(shí)際的分離操作帶來(lái)一些麻煩。
2.2.2少數(shù)配位吸附樹脂的化學(xué)穩(wěn)定性差 如載Fe3+的樹脂在吸附苯胺時(shí),會(huì)將苯胺氧化成苯胺黑。這種物質(zhì)不溶于水,會(huì)使樹脂的孔被堵塞[14]。苯胺的氧化是由于Fe3+具有一定的氧化性,F(xiàn)e3+在氧化苯胺的同時(shí)變成Fe2+。
另外,有些金屬離子如Ag+和Ni2+等價(jià)格較昂貴,從而限制了它們的大規(guī)模應(yīng)用。
3 配位吸附樹脂的基體
負(fù)載金屬離子的樹脂稱為配位吸附樹脂的基體。不同的基體對(duì)金屬離子的結(jié)合能力不同。不同基體的配位吸附樹脂,在負(fù)載上同樣的金屬離子后,對(duì)同一種配體的吸附能力也會(huì)有一定的差異。
3.1 磺酸型樹脂基體
磺酸型的基體具有較高的機(jī)械強(qiáng)度,負(fù)載了金屬離子后對(duì)配體的吸附能力很強(qiáng),因?yàn)榛撬嵝蜆渲瑢?duì)金屬離子的結(jié)合僅靠靜電相互作用,被負(fù)載的金屬離子上的配位位點(diǎn)沒(méi)有被樹脂基體占據(jù),金屬離子的配位位點(diǎn)可全部用于配位吸附。但磺酸型基體的樹脂結(jié)合金屬離子的強(qiáng)度不高,被負(fù)載的金屬離子容易脫落。應(yīng)用實(shí)例有:載Al3+的磺酸型樹脂對(duì)DNA的分離[7];載Cu2+和Fe3+的磺酸型樹脂對(duì)多元醇和糖的分離[17];載Ni2+的磺酸型樹脂對(duì)乙醇胺的分離[19]等。
3.2 丙烯酸型樹脂基體
丙烯酸型基體結(jié)合金屬離子的強(qiáng)度要比磺酸型基體大得多,被負(fù)載的金屬離子不容易脫落。但這種基體的樹脂對(duì)配體的吸附能力比磺酸型基體的樹脂弱得多,因?yàn)轸然系难跖c被負(fù)載的金屬離子間有配位作用,從而占據(jù)一定的配位吸附的位點(diǎn)。另外丙烯酸型樹脂基體的機(jī)械強(qiáng)度比較差,在高效液相色譜中的應(yīng)用受到一定的限制。其實(shí)例有:載Cu2+的丙烯酸樹脂分離硫化物[20]和安非它明藥物[10];載Cu2+和Zn2+的丙烯酸樹脂分離二胺和多胺[18];載Ni2+的丙烯酸型樹脂分離乙醇胺;載Cu2+和Zn2+的丙烯酸樹脂對(duì)氨基酸的分離[5]等。
3.3 氨基乙酸螯合型樹脂基體
這種基體對(duì)金屬離子的結(jié)合非常強(qiáng),被負(fù)載的金屬很少脫落,可在較寬的pH 范圍內(nèi)使用。如載Cu2+的氨基乙酸樹脂對(duì)硒酸根的富集[34]和對(duì)嗎啡等生物堿的色譜分離[18];載Ni2+的氨基乙酸型樹脂對(duì)砷酸根和亞砷酸根的富集和回收[31]等。
3.4 鍵合手性氨基酸的樹脂基體
這種樹脂負(fù)載了金屬離子后,主要用于手性氨基酸的拆分。樹脂的骨架有聚苯乙烯類[21,22]、聚丙烯酰胺類[36]、聚丙烯酸酯類[37]、聚乙烯胺類和硅膠類[38]等。其中聚苯乙烯類具廣譜性,能對(duì)20多種手性氨基酸進(jìn)行拆分,但由于骨架親水性不太好,拆分過(guò)程緩慢。丙烯酰胺型骨架親水性好,明顯提高了拆分效率。丙烯酸酯類骨架親水性也較好,且結(jié)合Cu2+離子非常牢固,分離過(guò)程中幾乎無(wú)銅脫落。聚乙烯胺型骨架親水性最好,可在較短的時(shí)間完成拆分過(guò)程,且對(duì)芳香氨基酸的拆分效果尤佳。硅膠類基體的手性配體交換樹脂不僅分離的速度快,效率高,而且在多種氨基酸混合物的拆分方面,尤顯獨(dú)特性能[26]。
4 配位吸附樹脂上負(fù)載的離子
配位吸附樹脂上負(fù)載的離子主要有Cu2+、Ni2+、Co2+、Fe3+、Fe2+、Ag+、Cr3+、Cd2+、Zn2+和Al3+等。選擇何種離子,需考慮樹脂基體對(duì)該離子的結(jié)合強(qiáng)度及負(fù)載量、離子對(duì)被吸附配體的結(jié)合能力以及離子本身的穩(wěn)定性等多方面的因素。但目前樹脂上負(fù)載的離子對(duì)配位吸附的影響沒(méi)有詳細(xì)的文獻(xiàn)報(bào)道。配位吸附時(shí)選擇何種離子載于樹脂上主要是靠經(jīng)驗(yàn)確定的。
Cu2+被樹脂基體結(jié)合的強(qiáng)度高,而且結(jié)合到基體上后對(duì)配體的吸附能力也比較強(qiáng)[2,39]。同時(shí)它方便易得,性質(zhì)比較穩(wěn)定,載Cu2+的配位吸附樹脂在應(yīng)用研究方面的報(bào)道最多[2,3,9,10,21,22]。
載Ni2+的配位吸附樹脂主要用于乙醇胺[19]、苯丙胺類藥物[10]和脂肪酸[16]的分離。通常磺酸型樹脂結(jié)合金屬離子的強(qiáng)度比其它類型基體的樹脂要弱,但它對(duì)Ni2+的結(jié)合強(qiáng)度卻非常高[39]。
載Fe3+的配位吸附樹脂主要集中在廢水處理和污染物的控制方面。如載Fe3+的配位吸附樹脂對(duì)廢水或海水中砷化物的富集和回收[29~32]、載Fe3+的磺酸型樹脂對(duì)芳胺和酚類物質(zhì)的去除和回收[14,15]、載Fe3+的螯合型樹脂對(duì)異硫氰酸鹽的去除和回收[35]等。也有少量載Fe3+的配位吸附樹脂在色譜分離上應(yīng)用的報(bào)道,如載Fe3+磺酸型樹脂對(duì)糖和多元醇的色譜分離[17]等。Fe3+的穩(wěn)定性較差,遇還原性配體時(shí)易被還原成Fe2+。如以載Fe3+的配位吸附樹脂吸附苯胺時(shí),苯胺被氧化成難溶于水的苯胺黑并附著在樹脂上,堵塞了樹脂的微孔,因此載Fe3+的配位吸附樹脂在應(yīng)用上受到限制,只能吸附化學(xué)性質(zhì)很穩(wěn)定的配體。載Fe3+的配位吸附樹脂以堿性的洗脫液洗脫時(shí),F(xiàn)e3+會(huì)變成Fe(OH)3 凝膠而牢固地被附著在樹脂上,而Fe(OH)3 凝膠對(duì)配體同樣具有較強(qiáng)吸附能力,不會(huì)影響樹脂的使用性能,而且隨著時(shí)間的延長(zhǎng),樹脂的吸附能力反而增強(qiáng)[29~31,34]。到最后Fe(OH)3 凝膠在樹脂上附著太多才使吸附量減小,這時(shí)需要用酸將Fe(OH)3 凝膠從樹脂上洗盡并以FeCl3 再生樹脂。由于Fe3+會(huì)使有的配體氧化,有時(shí)考慮用Fe2+載于樹脂上吸附相應(yīng)的配體,如載Fe2+的磺酸樹脂對(duì)芳胺的吸附[15]。Fe2+雖然不會(huì)氧化被吸附的配體,但Fe2+本身容易被空氣或水中的氧氧化成Fe3+,因而本身也不穩(wěn)定,必須在制備樹脂的當(dāng)日使用,因而載 Fe2+的配位吸附樹脂在應(yīng)用上受到更大的限制,報(bào)道很少。
載Co2+的配位吸附樹脂主要是將Co2+載在官能團(tuán)化的多孔花粉素上,用以分離芳胺及核苷酸和核酸[6,11,12]。載有其它幾種金屬離子的配位吸附樹脂僅見個(gè)別報(bào)道,如載Al3+的磺酸型樹脂分離DNA[7,8]、載Cd3+的硅膠分離芳胺異構(gòu)體[13]、載Zn2+的配位吸附樹脂分離二胺和多胺[18]、載Cr3+的配位吸附樹脂富集檸檬酸和酒石酸[32]、載Ag+的配位吸附樹脂吸附二胺和哌啶[3]等。
5 配位吸附的選擇性
配位吸附樹脂對(duì)不同配體的吸附選擇性主要取決于以下四個(gè)因素。
5.1 空間位阻[40]
對(duì)最常見的配體胺類而言,伯胺被吸附的強(qiáng)度最大,仲胺次之,叔胺最小。這是由配位吸附時(shí)空間位阻的不同造成的。另外,空間位阻的影響不僅取決于氮上取代基的大小和數(shù)量,還取決于與氮鄰近的碳上取代基的數(shù)目。
5.2 配體的堿性
通常配體的堿性越大,被吸附的強(qiáng)度也越大。如考察不同堿性的取代苯胺在載Cd 硅膠柱上的容量因子,發(fā)現(xiàn)堿性越大的取代苯胺在載Cd硅膠柱上保留時(shí)間越長(zhǎng)[13]。
5.3 空間構(gòu)象
在配體交換樹脂對(duì)手性氨基酸的拆分中,就是利用不同空間構(gòu)象的氨基酸在手性配體交換樹脂上被吸附的強(qiáng)度不同來(lái)進(jìn)行的(詳見7.2)。另外,在載Ca2+的配位吸附樹脂分離糖類化合物時(shí),樹脂對(duì)不同糖的吸附強(qiáng)度與糖分子上的羥基在單糖單元環(huán)上的不同構(gòu)象有關(guān)。Goulding 證實(shí),糖的羥基以a-e-a 鍵形式結(jié)合在環(huán)上,更易與Ca2+配位,具這種構(gòu)型的糖其吸附強(qiáng)度更大[40]。
5.4 配體分子中所含的配位原子數(shù)
在配位吸附樹脂對(duì)多胺的分離中,配體含氮原子數(shù)越多則吸附得越牢固[18]。
5.5 介質(zhì)的pH
在載Fe2+的配位吸附樹脂對(duì)幾種芳胺的吸附中,酸性介質(zhì)有利于配位吸附。這是因?yàn)樵谒嵝越橘|(zhì)中,F(xiàn)e2+易呈離子狀態(tài),這對(duì)配位吸附更為有利[15]。
6 配位吸附的熱力學(xué)和動(dòng)力學(xué)
6.1 熱力學(xué)
對(duì)于一般的吸附,其熱力學(xué)方程主要有Langmuir 吸附方程(C/q= C/qm+1/Kdqm,其中C為吸附質(zhì)平衡濃度,q為樹脂的吸附量,qm為樹脂的飽和吸附量,Kd為鍵合常數(shù))和Freundlich 吸附方程(q=kC1/n,其中k和n均為常數(shù))。這兩個(gè)方程對(duì)配位吸附也基本適用。尤其是Langmuir 吸附方程更能準(zhǔn)確地描述配位吸附的平衡[11,15,29~31]。
Chanda等曾推導(dǎo)了針對(duì)配位吸附的平衡方程[14,15,34],即:
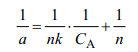
可以看出,這個(gè)方程與Langmuir吸附方程有完全相同的形式,但各項(xiàng)參數(shù)的意義不同,其中a為樹脂上每mol金屬離子吸附配體的數(shù)量,n為每mol金屬離子吸附配體的飽和吸附量,CA為樹脂的飽和吸附量,k為配位鍵合常數(shù)。以1/ a 對(duì)1/ CA作圖,可直接求出樹脂上每mol金屬離子吸附配體的飽和吸附量及吸附的配位鍵合常數(shù)k[14,34]。如載Fe3+的配位吸附樹脂在吸附SCN-時(shí),樹脂上的每個(gè)Fe3+可定量地與三個(gè)分子的SCN-結(jié)合[34]。
6.2 動(dòng)力學(xué)
對(duì)于一般的吸附,其動(dòng)力學(xué)過(guò)程與離子交換的動(dòng)力學(xué)過(guò)程基本相似,吸附的動(dòng)力學(xué)模型和方程基本上是在離子交換動(dòng)力學(xué)的基礎(chǔ)上作了一定的修正。離子交換的動(dòng)力學(xué)過(guò)程在很早以前就被深入地研究過(guò)。一般認(rèn)為離子的擴(kuò)散過(guò)程是離子交換的控速步驟( 包括膜擴(kuò)散和粒擴(kuò)散)[41]。許多研究者,如Boyd[41]、Levenspiel[42]、Helfferich[43]和Kressman[44]等都提出過(guò)判斷速率控制過(guò)程的方程或方法,其中Helfferich提出的膜擴(kuò)散/ 粒擴(kuò)散的簡(jiǎn)單判斷方法[43](即考察不同初始濃度的F(t )- t曲線。如曲線基本重合則為粒擴(kuò)散,如曲線有差異,即有初始濃度依賴性,則為膜擴(kuò)散)和Kressman 提出的判斷膜擴(kuò)散/ 粒擴(kuò)散控制的“間斷法”因簡(jiǎn)單易行,被廣泛應(yīng)用[14,29,30,34]。其它方法因涉及的參數(shù)太多,有的參數(shù)只是估計(jì)的近似值,或?qū)嶒?yàn)條件與建立方程的條件有差別,只能作為判斷的參考方法,用得不多。對(duì)于大多數(shù)配位吸附來(lái)說(shuō),粒擴(kuò)散是主要的控制步驟,如載Fe3+的配位吸附樹脂對(duì)砷化物[29,30]和酚類[14]的吸附,載Co2+的配位吸附樹脂對(duì)芳胺的吸附[11]等。但也有少數(shù)膜擴(kuò)散控制配位吸附的報(bào)道,如載Fe3+的配位吸附樹脂對(duì)硫氰酸鹽的吸附[34]等。
7 配位吸附的應(yīng)用
7.1 色譜方面的應(yīng)用
配位吸附的應(yīng)用主要在配體交換色譜方面,這方面的應(yīng)用研究報(bào)道最多,被分離的物質(zhì)種類繁多,如生物堿[9,10]、其它胺類[10~15,18,19]、醇和糖[17]、脂肪酸[16]、氨基酸[5]、硫化物[20]及核酸和核苷酸[6~8]等。這些被分離的物質(zhì)依靠分子中的氮、氧或硫原子與載體上的金屬離子配位,并利用其空間位阻、堿性和配位原子數(shù)目等因素的差異來(lái)進(jìn)行分離。配體交換色譜的出現(xiàn)使原來(lái)難以分離的許多化合物很容易分開,因而大大豐富了色譜分離的內(nèi)容。和常規(guī)色譜分離相比,配體交換色譜具有分離選擇性高、成本低、速度快的特點(diǎn)。配體交換色譜在分析上雖有獨(dú)特的優(yōu)勢(shì),但在色譜制備上存在明顯的不足。因?yàn)檩d體上的金屬離子會(huì)或多或少地脫落,分離的產(chǎn)物需進(jìn)一步純化,以去除混雜的金屬離子。
配體交換色譜以液相色譜為主,也可以用氣相色譜或薄層色譜的方式進(jìn)行分離。
7.2 配位吸附樹脂對(duì)手性氨基酸異構(gòu)體的拆分
手性配體交換色譜仍屬配體交換色譜的范疇,但由于其機(jī)理和應(yīng)用獨(dú)特,故在這里單獨(dú)介紹。手性配體交換色譜主要用于手性氨基酸異構(gòu)體的拆分。Davankov首先開創(chuàng)了將這一領(lǐng)域,30多年來(lái),它一直是手性氨基酸異構(gòu)體拆分的主要方法。手性配體交換樹脂對(duì)氨基酸對(duì)映體的吸附強(qiáng)度的差異與樹脂上擔(dān)載的手性氨基酸有直接的關(guān)系。Davankov[23]提出手性配體交換樹脂識(shí)別氨基酸對(duì)映體的模型,用以解釋手性配體交換樹脂對(duì)手性氨基酸對(duì)映體的識(shí)別機(jī)理。不管是哪種模型,有一點(diǎn)可以肯定,就是手性氨基酸的一對(duì)映體與樹脂上的Cu2+配位穩(wěn)定,而另一種對(duì)映體則因?yàn)榭臻g上的擁擠而不太穩(wěn)定。這種配位穩(wěn)定性的差異決定了手性氨基酸的不同異構(gòu)體在配位吸附樹脂上吸附強(qiáng)度的不同,從而很容易通過(guò)色譜的方法將它們分離開。傳統(tǒng)的物理法、化學(xué)法以及酶法等拆分氨基酸對(duì)映體的缺陷是效率低,需多次重復(fù)操作,且所用的手性制劑及酶制劑一般難以得到,不便回收及反復(fù)使用。采用通常的氣相色譜法和液相色譜法拆分,一般需要柱前樣品衍生化,費(fèi)時(shí)麻煩,效率低;而手性配體交換色譜可直接拆分氨基酸對(duì)映體,無(wú)需進(jìn)行柱前衍生化,分離速度快,且選擇性高,流動(dòng)相多采用水,實(shí)驗(yàn)成本低。目前手性配體固定相多采用在硅膠上鍵合手性分子再載以金屬離子來(lái)制備,使之既具有較強(qiáng)的機(jī)械強(qiáng)度,又具有適當(dāng)?shù)挠H水性,以提高色譜分離的速度。手性配體交換色譜在色譜制備上同樣存在不足。除上述金屬離子脫落的原因外,手性配體交換色譜的固定相對(duì)樣品的處理量不大,也大大影響制備的效率。因?yàn)樵谳d體上擔(dān)載手性分子后,載體的比表面和孔容都將大大減小。如比表面為334.1m2/g,孔容為0.82mL/g 的硅膠,枝接上L-羥脯胺酸后比表面減至154.6m2/g,孔容減至0.40mL/g。比表面和孔容的減小將大大減少對(duì)硅膠樣品的處理量。
7.3 廢水處理及污染物的控制
水處理是配位吸附應(yīng)用的重要領(lǐng)域之一。由于配位吸附具有很高的選擇性,它可以有效地去除其它方法難以去除的水污染物[45~47]。一種通過(guò)賴氨酸臂將兩個(gè)乙酸基連接在聚苯乙烯骨架上的螯合樹脂[31],絡(luò)合 Fe3+的強(qiáng)度非常高,能在極寬的 pH 范圍內(nèi)使用。這種樹脂對(duì)廢水中的污染物砷酸根及亞砷酸根具有很強(qiáng)富集性能。在 pH為3.5 時(shí)對(duì)砷酸根的吸附量為0.74mmol/g ,在pH為9 時(shí)對(duì)亞砷酸的吸附量為0.84mmol/g。由于對(duì)這兩種不同價(jià)態(tài)砷化物吸附的最大pH 不同,可將五價(jià)砷和三價(jià)砷分開。樹脂可用0.1mol/L 的NaOH再生,只有0.1%的Fe3+從樹脂上泄漏,而兩種砷化物可完全被洗脫。另外,載Fe3+的XFS-4195樹脂、載 Fe3+的Chelex100樹脂及載Fe3+的UR-10螯合樹脂等,均能很好地吸附砷化物,能將海水中的微量砷富集100~200倍[28~30]。芳胺和酚類是另一類重要的污染物。這兩類污染物的去除已有大量的文獻(xiàn)報(bào)道,但配位吸附的方法是一種選擇性更高的方法。載Co2+的配位吸附樹脂對(duì)芳胺類化合物具有很好的吸附能力[11],載Fe2+的磺酸樹脂對(duì)各種芳胺均具良好的吸附性能,而且用稀鹽酸可有效地洗脫[15]。由原煤生產(chǎn)液化氣的工業(yè)廢水中含幾百ppm的硫氰酸鹽,它可被載Fe3+的螯合樹脂XFS-4195有效地去除,樹脂對(duì)SCN-的吸附量達(dá)75mg/g,而且很容易被稀堿液洗脫,洗脫時(shí)無(wú)Fe3+泄漏,樹脂可反復(fù)使用。
7.4 配位吸附樹脂對(duì)陰離子的富集和回收
雖然離子交換色譜可將各種離子分開,但要在工業(yè)規(guī)模上有選擇地富集和回收某些離子還有難度,尤其是在其它競(jìng)爭(zhēng)性離子濃度較高時(shí)。采用配位吸附的方法可能會(huì)有選擇地吸附和分離那些可與金屬離子配位的離子,如磷酸根和鄰苯二甲酸根等。但單純的配位吸附對(duì)這些離子型的配體不具有吸附分離效果,配位吸附通常只針對(duì)非離子型的物質(zhì)。因此,為了有選擇地吸附分離這類配體型的離子物質(zhì),需將配位吸附和離子交換兩者結(jié)合起來(lái),即將金屬離子擔(dān)載在螯合樹脂上( 而不是離子交換樹脂上),則擔(dān)載的金屬離子與待吸附的離子型配體間不僅存在配位作用,也存在正負(fù)電荷之間的作用,其中配位作用決定了樹脂對(duì)配體型離子的選擇作用,而正負(fù)電荷平衡得以使配位吸附能夠?qū)崿F(xiàn)[35]。采用這種方法可有效地在其它競(jìng)爭(zhēng)性離子濃度較高時(shí)有選擇性地吸附酒石酸、檸檬酸[32]和磷酸根[48]、砷酸根和亞砷酸根[33,35]等陰離子。采用堿-鹽混合溶液或酸-鹽混合溶液能有效地將被吸附的配體型離子洗脫下來(lái)。
8 配位吸附的發(fā)展趨勢(shì)
配位吸附的發(fā)展經(jīng)歷了近半個(gè)世紀(jì)的歷程,目前手性配體交換色譜仍是其應(yīng)用的主要領(lǐng)域。
這一領(lǐng)域未來(lái)的發(fā)展方向是,提高手性鍵合固定相中金屬離子的固載量,以提高色譜分離的效果,并使色譜分離向制備或半制備的方向發(fā)展;在載體上固載新型手性分子,以提高分離的選擇性,或降低手性固定相制備的成本。
配位吸附的材料目前主要被制成球形顆粒用作色譜分離的固定相或其它分離材料。它也可以被制成膜的形式,使其應(yīng)用領(lǐng)域得到進(jìn)一步擴(kuò)展。這方面的研究處于剛起步階段,如將L- 脯胺酸連接到交聯(lián)聚乙烯醇膜上,再載以銅離子,可有效地拆分多種手性氨基酸異構(gòu)體。近年來(lái)膜分離技術(shù)已越來(lái)越成熟,并已獲得了廣泛的應(yīng)用。隨著膜技術(shù)的發(fā)展,配位吸附材料未來(lái)可能會(huì)更多地被制成膜的形式,并在更多的領(lǐng)域內(nèi)發(fā)揮作用。
天然產(chǎn)物中有許多活性成分如生物堿、多酚類及多糖類,都具有配體性質(zhì),因而原則上可以采用配位吸附進(jìn)行分離。如一種載Zn2+的配位吸附樹脂能有效地吸附分離維生素B12。將銅離子載于磺酸樹脂上,能從干苔中分離純化干苔多糖。這種干苔多糖具有抗腫瘤活性,并能與固定化藻藍(lán)蛋白協(xié)同作用以提高其抑制癌細(xì)胞的效果。采用配位吸附從天然產(chǎn)物中分離活性成分的一大優(yōu)勢(shì)是吸附可以在有機(jī)介質(zhì)中進(jìn)行。大多生物活性成分在進(jìn)行吸附分離前均需經(jīng)有機(jī)溶劑提取。如采用常規(guī)吸附分離,需回收有機(jī)溶劑,使活性成分的有機(jī)溶液轉(zhuǎn)化成水溶液,這一轉(zhuǎn)化不僅增加了分離工藝的復(fù)雜性,更重要的是造成相當(dāng)部分的活性成分的損失。而配位吸附可以直接在有機(jī)介質(zhì)中進(jìn)行,因?yàn)榕湮晃酵ǔ2粫?huì)被有機(jī)介質(zhì)所抑制[49]。另外,有機(jī)介質(zhì)中的配位吸附還能有效地防止金屬離子的脫落。隨著非水介質(zhì)中配位吸附研究的不斷發(fā)展,配位吸附可望在天然產(chǎn)物分離方面得到進(jìn)一步的應(yīng)用。
參考文獻(xiàn)
[1] F Helfferich. Nature, 1961, 189: 1001~1002.
[2] H F Walton. Ind. Eng. Chem. Res., 1995, 34: 2553~2556.
[3] R H Stokes, H F Walton. J. Am. Chem. Soc., 1954, 76: 3327~3330.
[4] F Helfferich. J. Am. Ch em. Soc., 1962, 84: 3237~3242.
[5] M Doury-Berthod, C Poitrenaud, B Tremillon. J. Chromatogr., 1979, 179: 37~39.
[6] E Pehlivan, S Yildiz. Sep. Sci. Technol., 1994, 29: 887~889.
[7] R M Kothri. J. Chromatogr., 1970, 52: 119~123.
[8] R M Kothri. J. Chromatogr.,1971, 56: 151~156.
[9] E Murgia, H F Walton. J. Chromatogr., 1975, 104: 417~419.
[10] C M Hernandez, H F Walton. Anal. Chem., 1972, 44(6): 890~894.
[11] M Ersoz, U S Vural, S Yildiz. Sep. Sci. Technol., 1995, 30: 3555~3557.
[12] E Pehlivan, U S Vural, A Ayar et al. Sep. Sci. Technol., 1996, 31(11): 1643~1646.
[13] D Kunzru, R W Frei. J. Chromatogr. Sci., 1974, 12: 191~196.
[14] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1983, 1: 183~189.
[15] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym. 1984, 2: 279~285.
[16 R Bedetti, V Carunchio, A Marino. J. Chromatogr., 1974, 95: 127~129.
[17] V Shaw, H FWalton. J. Chromatogr., 1972, 68: 267~269.
[18] J D Navratil, H F Walton. Anal. Chem., 1975, 47: 2443~2446.
[19] K Shimomura, T J Hsu, H F Walton. Anal. Chem., 1973, 45(3): 501~505.
[20] J W Vogh, J Edooley. Anal. Chem., 1975, 47: 816~818.
[21] V A Davankov, S V Rogozhin. J. Chromatogr., 1971, 60: 280~283.
[22] V A Davankov, S V Rogozhin, A V Semechkin et al. J. Chromatogr., 1973, 82: 359~364.
[23] V A Davankov, Y A Zolotarew. J. Chromatogr., 1978, 155: 285~288.
[24] Y A Zoloterew, N F Myasoedov, V A Penkina. J. Chromatogr., 1981, 207: 231~235.
[25] V A Davankov, S V Rogozhin, A V Semechkin et al. J. Chromatogr., 1974, 93: 363~366.
[26] D Charmot, R Audebert, C Quivoron. Liq. Chromatogr., 1985, 8: 1769~1774.
[27] D Charmot, R Audebert, C Quivoron. Liq. Chromatogr., 1985, 8: 1753~1768.
[28] I Yoshida, K Ueno, H Kobayashi. Sep. Sci. Technol., 1978, 13: 173~176.
[29] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1988, 7: 251~255.
[30] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1988, 8: 85~89.
[31] H Matsunaga, T Yokoyama, R J Edridge et al. React. Funct. Polym., 1996, 29: 167~171.
[32] Z Matajka, R Weber. React. Funct. Polym., 1990, 13: 299~302.
[33] D Zhao, A KSengupta, Y Zhu. Ind. Eng. Chem. Res., 1995, 34: 2676~2680.
[34] M Chanda, K F O’driscoll, G L Rempel. React. Funct. Polym., 1984, 2: 175~181.
[35] Y Zhu, A K Sengupta, A Ramana. React. Funct. Polym., 1992, 17: 229~234.
[36] R Audebert. J.Liq.Chromatogr., 1979, 2: 1063~1067.
[37] I A Yamskov, B B Berezin, V A Davankov. J. Chromatogr., 1981, 217: 539~543.
[38] R Audebert. J. Liq. Chromatogr., 1981, 4: 185~192.
[39] H F Walton. J. Chromatogr., 1974, 102: 57~59.
[40] R W Goulding. J. Chromatogr., 1975, 103: 229~232.
[41] G E Boyd, A W Adamson, L S Myers. J. Am. Chem. Soc., 1947, 69: 2836~2841.
[42] O Levenspiel. Chem. React. Eng.. New York: John Wiley, 1962: 343~344.
[43] F Helffrich. Ion Exchange. New York: McGrow-Hill, 1962: 324~326.
[44] T R Kressman. J A Kitcheher. Discuss. Faraday Soc., 1949, 7: 90~95.
[45] M Ersoz, M Yigitoglu, A Ayar. J. Appl. Polym. Sci., 1997, 64: 1225~1231.
[46] D Zhao, A K Sengupta. Ind. Eng. Chem. Res., 2000, 39(2): 455~462.
[47] D Zhao, A K Sengupta, L Stewart. Ind. Eng. Chem. Res., 1998, 37(11): 4383~4387.
[48] D Zhao, A K Sengupta. Water Res., 1998, 32(5): 1613~1625.
[49] J Z Li, Z Q Shi, YG Fan. Chinese J. Polym. Sci., 2002, 21: 439~444.
注:本文為提供者整理翻譯的,由于知識(shí)所限,錯(cuò)誤在所難免,敬請(qǐng)?jiān)彙H缬袉?wèn)題可以查找原文。
統(tǒng)計(jì)數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)
-
物竟數(shù)據(jù)庫(kù) 手機(jī)版
國(guó)內(nèi)首款化工類專業(yè)手機(jī)應(yīng)用
-
微博賬號(hào)
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競(jìng)
物競(jìng)數(shù)據(jù)庫(kù)是一個(gè)全面、專業(yè)、專注,并且免費(fèi)的中文化學(xué)品信息庫(kù),為學(xué)生、學(xué)者、化學(xué)品研究機(jī)構(gòu)、檢測(cè)機(jī)構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺(tái)進(jìn)行交流。
數(shù)據(jù)庫(kù)采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國(guó)內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號(hào):物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)

-
微博賬號(hào):wjhxp
聯(lián)系我們
上海市延長(zhǎng)路149號(hào)上海大學(xué)科技園412室
公司總機(jī): 021-56389801
訂購(gòu)電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競(jìng)數(shù)據(jù)庫(kù) 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫(kù) | 關(guān)于物競(jìng) | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號(hào)
滬公網(wǎng)安備 31010602001115號(hào)