匹伐他汀鈣 Pitavastatin calcium

結(jié)構(gòu)式
| 物競(jìng)編號(hào) | 0A8Y |
|---|---|
| 分子式 | (C25H23FNO4)2.Ca |
| 分子量 | 880.98 |
| 標(biāo)簽 | Livalo, 抑制劑 |
編號(hào)系統(tǒng)
CAS號(hào):147526-32-7
MDL號(hào):暫無
EINECS號(hào):暫無
RTECS號(hào):暫無
BRN號(hào):暫無
PubChem號(hào):暫無
物性數(shù)據(jù)
性狀:白色固體。
毒理學(xué)數(shù)據(jù)
暫無
生態(tài)學(xué)數(shù)據(jù)
暫無
分子結(jié)構(gòu)數(shù)據(jù)
暫無
計(jì)算化學(xué)數(shù)據(jù)
1.疏水參數(shù)計(jì)算參考值(XlogP):無
2.氫鍵供體數(shù)量:4
3.氫鍵受體數(shù)量:12
4.可旋轉(zhuǎn)化學(xué)鍵數(shù)量:14
5.互變異構(gòu)體數(shù)量:無
6.拓?fù)浞肿訕O性表面積187
7.重原子數(shù)量:63
8.表面電荷:0
9.復(fù)雜度:626
10.同位素原子數(shù)量:0
11.確定原子立構(gòu)中心數(shù)量:4
12.不確定原子立構(gòu)中心數(shù)量:0
13.確定化學(xué)鍵立構(gòu)中心數(shù)量:2
14.不確定化學(xué)鍵立構(gòu)中心數(shù)量:0
15.共價(jià)鍵單元數(shù)量:3
性質(zhì)與穩(wěn)定性
暫無
貯存方法
暫無
合成方法
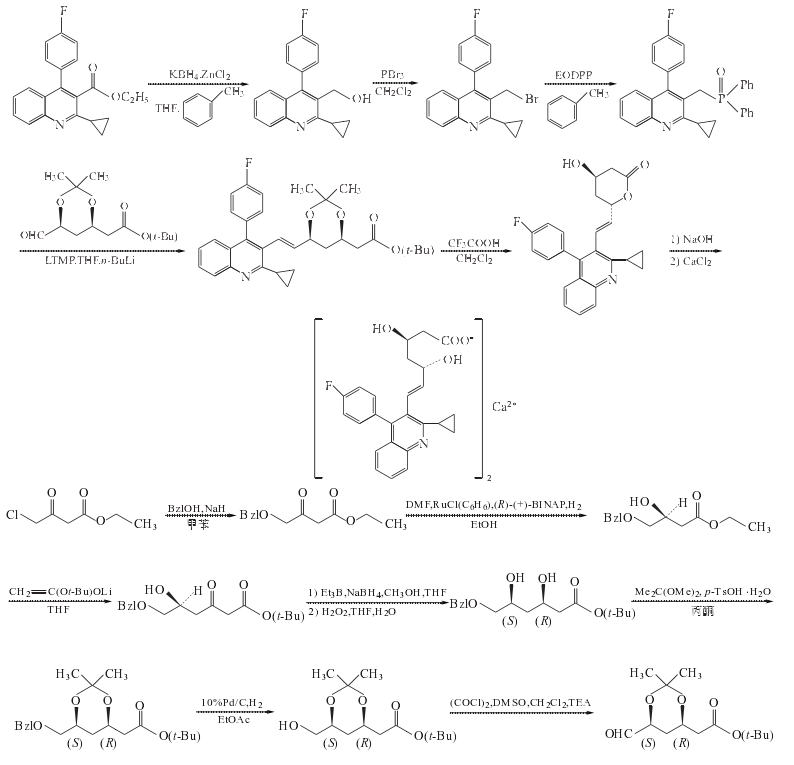
1. 2-環(huán)丙基-4-(4-氟苯基)喹啉-3-甲醇的制備
在反應(yīng)瓶中加入THF196ml、ZnCl2 31.5g(0.23mol)和KBH4 24.5g(0.45mol),室溫?cái)嚢?h.加入2-環(huán)丙基-4-(4-氟苯基)喹啉-3-羧酸乙酯49g(0.15mol)的甲苯392ml溶液,緩慢升溫至回流,攪拌回流反應(yīng)20h.冷至室溫,過濾,濾餅用甲苯加熱回流洗滌,抽濾,合并有機(jī)層,加水后靜置分層,水層用甲苯提取,合并有機(jī)層,用0.1mol/LNaOH水溶液洗滌,飽和鹽水洗至中性.減壓濃縮至干,剩余物用甲苯重結(jié)晶,得2-環(huán)丙基-4-(4-氟苯基)喹啉-3-甲醇27.7g,收率65%,mp128~130oC(文獻(xiàn)[4]收率70%,mp136~137oC)
2. 3-溴甲基-2-環(huán)丙基-4-(4-氟苯基)喹啉的制備
在反應(yīng)瓶中加入上步制備的化合物2-環(huán)丙基-4-(4-氟苯基)喹啉-3-甲醇25g(85mmol)和二氯甲烷200ml,降溫至0oC,在0oC攪拌滴加三溴化磷(PBr3)16ml(170mmol)的二氯甲烷50ml溶液,溫?cái)嚢璺磻?yīng)3h.蒸去二氯甲烷,余物緩慢倒入飽和NaHCO3水溶液中,至pH8~9,乙酸乙酯提取,機(jī)相用水洗至中性,壓濃縮至干,余物用無水乙醇重結(jié)晶,3-溴甲基-2-環(huán)丙基-4-(4-氟苯基)喹啉27g,收率90%mp129~131oC(文獻(xiàn)[6]收率89%,mp140 oC)
3. 2-環(huán)丙基-3-(二苯基氧膦甲基)-4-(4-氟苯基)喹啉的制備
在茄形瓶中加入上步制備的化合物3-溴甲基-2-環(huán)丙基-4-(4-氟苯基)喹啉27g(75.8mmol)、乙氧基二苯基膦35ml(150mmol)和甲苯270ml,回流12h.減壓蒸干,剩余物用二氯甲烷/石油醚重結(jié)晶,得2-環(huán)丙基-3-(二苯基氧膦甲基)-4-(4-氟苯基)喹啉36.0g,收率定量,mp172~174oC.
4. (3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯的制備
(1) 芐氧基乙酰乙酸乙酯的制備
在反應(yīng)瓶中加入甲苯1000ml和55%~60%NaH73.5g(1.68~1.84mol),攪拌懸浮,降溫至20~25oC,往懸浮液滴加苯甲醇162g(155ml,1.498mol),在50min內(nèi)滴加完,滴加過程中應(yīng)用冰水浴微微冷卻以保持溫度在20~25oC范圍內(nèi),滴畢,將稠厚的漿狀液于24oC繼續(xù)攪拌2h,氫氣逸出終止.再在20~25oC攪拌滴加4-氯乙酰乙酸乙酯125g,(103ml,1.7595mol),30min內(nèi)滴加完,滴加過程中因放熱反應(yīng),要適度冷卻,以保持溫度恒定在20~25oC之間.滴畢,繼續(xù)在03M攪拌反應(yīng)19h.反應(yīng)過程中用GC進(jìn)行監(jiān)測(cè)(GCl,tR:苯甲醇1.2min,化合物4-氯乙酰乙酸乙酯2.0min,化合物芐氧基乙酰乙酸乙酯7.1min)反應(yīng)畢,于20~25oC并微冷卻下,在30min內(nèi)加入2mol/L檸檬酸水溶液755ml(1.51mol),當(dāng)該溶液加入30%以后,反應(yīng)液成為非常稠厚漿狀物,繼續(xù)加入后,則反應(yīng)液黏度降低.最后反應(yīng)混合物pH為4.靜置分層,水溶液層用甲苯100ml提取,合并有機(jī)層,用無水Na2SO4干燥,過濾,濾液減壓蒸除溶劑,剩余油狀物加庚烷(25ml×2)一起攪拌,分取庚烷層,仍為油狀的庚烷層真空下濃縮,得粗品225g,將該粗品用雙級(jí)薄膜蒸發(fā)器進(jìn)行真空蒸餾[%控制第一蒸餾柱溫140oC,第二蒸餾柱溫180oC,真空度為66.66Pa(0.5Torr),得芐氧基乙酰乙酸乙酯純品150g,收率84%,為無色黏滯油狀物#純度>99%
(2)(S)4-芐氧基-3-羥基丁酸乙酯的制備
在反應(yīng)瓶中加入新鮮蒸餾的DMF900ml,在氬氣通過液面以下保護(hù)下再加入(R)-(+)-2,2’-雙(二苯膦基)-1,1’-聯(lián)萘和苯基氯化釕二聚物7.0g(14.0mmol,28.0mmol RU),在氬氣連續(xù)通過混合物的條件下,攪拌15min.然后反應(yīng)瓶深深浸入預(yù)先加熱到100oC的油浴中,則溶液澄清,并變成紅棕色清液.將其在氬氣保護(hù)下冷至25oC備用.此時(shí),在30L不銹鋼高壓反應(yīng)釜中加入無水乙醇10L和上步反應(yīng)制備的化合物芐氧基乙酰乙酸乙酯保持釜內(nèi)微帶正壓的氬氣保護(hù)下加入上述制備的催化劑,加畢,密閉高壓釜,用H2置換釜中氬氣3次.然后充H2,攪拌加熱至內(nèi)溫100oC此時(shí)再通入H2調(diào)釜內(nèi)H2壓力為0.4MPa,恒壓0.4MPa H2壓力和100oC下,一般反應(yīng)12H則達(dá)反應(yīng)終點(diǎn).反應(yīng)畢,降至室溫,通過取樣閥取小樣進(jìn)行分析,GC3分析表明>99%已氫化.HPLC分析粗品的光學(xué)純度為97.4%.用N2將釜內(nèi)氣體清除3次,反應(yīng)液放至蒸餾釜,高壓釜用乙醇1L進(jìn)行清洗,洗液與反應(yīng)液合并,真空蒸除乙醇,高真空60oC下蒸除DMF.剩余物用薄膜蒸發(fā)器于140~150oC蒸餾柱溫/13.33Pa條件下蒸出目的物(S)4-芐氧基-3-羥基丁酸乙酯,得(S)4-芐氧基-3-羥基丁酸乙酯9.70kg,收率96%,為無色油狀物#如果在0~10oC則固化結(jié)晶.
(3)(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯的制備
在反應(yīng)釜中加入二異丙胺9.4L,(71.7mol)和THF13.4L,攪拌下于0~10oC1h內(nèi)加入15%丁基鋰的己烷溶液43.6L(69.8mol),該混合溶液在10oC攪拌30min,再于-40oC 40min內(nèi)滴加乙酸叔丁酯9.0ml(66.8mol),再于-40oC 30min內(nèi)滴加上步制備的化合物(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯4.0kg(16.8mol)的THF4L溶液,將混合物于-40oC攪拌反應(yīng)1h.用TLC跟蹤確認(rèn)反應(yīng)達(dá)終點(diǎn)后,往反應(yīng)混合物中加水(沒有冷卻)7.4L,30min加完,加完水后使反應(yīng)混合物達(dá)-10oC分出有機(jī)層,水層用甲苯(16L×2)提取,合并有機(jī)層,用無水Na2SO4干燥,過濾,濾液減壓蒸除溶劑[,得半固體粗品(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯7.3~7.7kg,內(nèi)含有相當(dāng)量的溶劑.
將粗品(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯的半固體用異丙醚洗滌研磨搗碎后過濾,高真空下干燥,可得純的分析試樣,mp136~137oC.
(4)3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯的制備
在反應(yīng)釜中加入THF7.5L和上步制備的化合物(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯7.54kg(16.8mol),攪拌溶解,20~25oC15min內(nèi)加入1mol/L三乙基硼的己烷溶液40.0L(40.0mol),反應(yīng)吸熱.加畢,將溶液攪拌10min.然后冷卻至-60oC,立刻加入硼氫化鈉(NaBH4)1.9kg(50.4mol)(反應(yīng)不放熱),再在-60oC 2.5h內(nèi)滴加甲醇35L(強(qiáng)放熱),滴加畢,在同溫度下攪拌1h.TLC跟蹤[展開劑為庚烷/乙酸乙酯(2:1)]確認(rèn)反應(yīng)終點(diǎn),反應(yīng)畢,15min內(nèi)加水17L,將反應(yīng)混合物轉(zhuǎn)至裝有攪拌器的200L分離器中,加入鹽水17L,分相,水相用甲苯(33L×2)提取,分相后沉淀鹽被沉積在分離器底部.合并有機(jī)相,用水22L洗滌#然后加入無水Na2SO4過夜.過濾掉干燥劑Na2SO4后,將濾液真空濃縮.剩余物加THF 34L溶解,在0~10 oC和有效冷卻下加入35%H2O2 7.5L,在1h內(nèi)加完.將混合物繼續(xù)攪拌30min,TLC跟蹤,確認(rèn)中間體硼酯反應(yīng)完全,并形成了化合物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯后,加入水16L和甲苯16L.再將混合物攪拌5min,靜置分相.在有效冷卻下,往分出的有機(jī)相加入NaHCO3 2.0kg,破壞過量的H2O2.混合物再攪拌10min.有機(jī)相用飽和的NaHCO3水溶液10L洗滌,然后用鹽水5L洗滌,無水Na2SO4干燥,過濾,濾液減壓蒸除溶劑,得油狀剩余物7.0kg,與庚烷12L混合后轉(zhuǎn)至裝有攪拌器的100L分離器中,將混合物強(qiáng)烈攪拌1h.然后讓其沉降2~3h.從產(chǎn)物相分取庚烷相,庚烷相再加入庚烷24L稀釋,于5 oC攪拌過夜.分取油狀產(chǎn)物相,含在合并產(chǎn)物相的殘留溶劑減壓蒸除,得純的3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯4.2kg(13.5mol),以(S)4-芐氧基-3-羥基丁酸乙酯計(jì)收率80%,產(chǎn)物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯為特別淡的黃色油狀物.
(5)(3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯的制備
在反應(yīng)釜中加入上步制備的化合物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯11.1kg(35.76mol)和丙酮112L,開攪拌,冷至20~25oC,加入2,2-二甲氧基丙烷7.35kg(8.68L,69.2mol)和對(duì)甲苯磺酸一水合物0.68kg(3.6mol),將起初的無色溶液于24oC攪拌4h.在攪拌1~2h后溶液慢慢轉(zhuǎn)變成黃色,用HLC跟蹤,確認(rèn)化合物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯已反應(yīng)生成產(chǎn)物(3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯后,加入固體NaHCO3 2.4kg(28.6mol),將懸浮液攪拌30min.然后將混合物過濾,濾液真空蒸除溶劑,得無色油狀物11.5kg,粗品收率92%.
往200cm×30cm(外徑)(體積141L)MPLC柱裝入硅膠(70~210um)110kg和環(huán)己烷/乙酸乙酯(8:1)200L配成的漿料(溢流裝柱).將上述粗品11.5kg溶于環(huán)己烷5L中,經(jīng)過濾除去甲苯磺酸鈉后的濾液進(jìn)入上述色譜柱分離純化(詳細(xì)操作見文獻(xiàn)[9]),經(jīng)標(biāo)準(zhǔn)后處理和高真空干燥后,得純的化合物(3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯6.9kg,收率69%.純度>99%(HPLC),產(chǎn)物為無色油狀物.
(6) (3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯的制備
在40L的搪瓷高壓釜中加入上步制備的化合物3.25kg(9.27mol)和乙酸乙酯30L,用N2置換溶液中和釜中空氣,再加入Pd/C(10%)催化劑0.32kg,用H2置換釜中N2后充H2(壓力1MPa),攪拌下和恒氫氣壓力(1MPa)下于31~33氫化反應(yīng)5h,經(jīng)取樣閥取試樣進(jìn)行TLC分析[展開劑:庚烷/乙酸乙酯(1:1)](3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯斑點(diǎn)消失,產(chǎn)物(3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯斑點(diǎn)明顯,則反應(yīng)達(dá)終點(diǎn).用N2清除釜內(nèi)氫氣,過濾,濾液真空濃縮蒸除溶劑,剩余物加甲苯(1L×2),每次都真空蒸除甲苯,剩余物在高真空下干燥,得黏滯無色油狀物(3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯2.42kg,收率100%,純度>98%.
(7) (3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯的制備
在干燥的四口反應(yīng)瓶中加入干燥的二氯甲烷25ml和草酰氯1.0ml(11mmol),降溫至-50~-60oC,攪拌下慢慢往草酰氯的二氯甲烷溶液滴加DMSO1.7ml(1.72g,22mmol)與二氯甲烷5ml混合液,反應(yīng)混合物攪拌2min,然后在同溫度下5min內(nèi)滴加上步制備的化合物(3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯2.6g(10mmol)與二氯甲烷10ml的混合液,加畢,連續(xù)攪拌15min.再加入三乙胺,將混合物攪拌5min.然后升至室溫#加水50ml,攪拌片刻后靜置分層,水層用二氯甲烷50ml反提取.合并有機(jī)層,用飽和Na2CO3水溶液洗滌,無水MgSO4干燥,過濾,濾液用旋轉(zhuǎn)蒸發(fā)器濃縮至25ml,依次用1%稀鹽酸、水、5%稀Na2CO3水溶液和水洗滌,然后將有機(jī)相濃縮至無溶劑,則得(3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯的粗品2.50g,收率97%,可直接用于下步反應(yīng).
5. (3R,5S,6E)-7-[2-環(huán)丙基-4-(4-氟苯基)喹啉-3-基]-3,5-二羥基-3,5-O-異亞丙基-6-庚烯酸叔丁酯的制備
在反應(yīng)瓶中加入2,2,6,6-四甲基哌啶鋰20ml(120mmol)和THF200ml,0oC和N2保護(hù)下,攪拌滴加2.5mol/L正丁基鋰,滴畢,于0oC攪拌15min.冷卻至-78oC,滴加化合物(3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯50g(105mmol)的THF800ml溶液,攪拌1h.加入上步制備的中間體(3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯33.5g(130mmol)的THF200ml溶液,攪拌1h后升至室溫反應(yīng)過夜.加入飽和NaHCO3水溶液500ml終止反應(yīng),用乙酸乙酯提取,提取相用飽和鹽水洗至中性,無水Na2SO4干燥,過濾,濾液減壓蒸干,剩余物經(jīng)硅膠柱色譜分離,得油狀物37.5g,收率69%.
6. (4R,6S,E)-6-[2-環(huán)丙基-4-(4-氟苯基)喹啉-3-基-乙烯基]羥基-3,4,5,6-四氫-2H-吡喃-2-酮的制備
在反應(yīng)瓶中加入上步制備的化合物(3R,5S,6E)-7-[2-環(huán)丙基-4-(4-氟苯基)喹啉-3-基]-3,5-二羥基-3,5-O-異亞丙基-6-庚烯酸叔丁酯2.52g(4.9mol)的二氯甲烷20ml溶液,攪拌降溫至0oC,滴加三氟乙酸5.6ml(73.5mmol)的二氯甲烷8ml溶液,滴畢,室溫?cái)嚢璺磻?yīng)2h.加入飽和NaHCO3水溶液終止反應(yīng).調(diào)至pH7~8,用二氯甲烷提取,提取相用水洗滌,無水Na2SO4干燥,過濾,濾液減壓濃縮至干,得1.97g,收率99.5%,mp128~130.
7. (3R,5S,6E)-(+)-7-[2-環(huán)丙基-4-(4-氟苯基)喹啉-3-基] -3,5-二羥基-6-庚烯酸鈣鹽(2:1)(匹伐他汀鈣)的合成
在反應(yīng)瓶中加入上步制備的化合物(4R,6S,E)-6-[2-環(huán)丙基-4-(4-氟苯基)喹啉-3-基-乙烯基]羥基-3,4,5,6-四氫-2H-吡喃-2-酮1.2g(3.0mmol)和無水乙醇20ml,攪拌溶解,再加入0.1mol/LNaOH水溶液32ml,室溫?cái)嚢璺磻?yīng)1h.加入0.1mol/L鹽酸調(diào)至pH7,減壓蒸除溶劑,剩余物中加水20ml和無水氯化鈣0.32g(2.9mmol),攪拌15min,后抽濾,濾餅用水洗滌,抽干,烘干,用異丙醇/水重結(jié)晶,得白色固體(3R,5S,6E)-(+)-7-[2-環(huán)丙基-4-(4-氟苯基)喹啉-3-基] -3,5-二羥基-6-庚烯酸鈣鹽(2:1)(匹伐他汀鈣)1.0g,收率76.3%.
用途
本品為3-羥基-3-甲基戊二酰輔酶A還原酶抑制劑,可以減少肝臟合成膽固醇,使血漿低密度脂蛋白膽固醇的清除增加,可阻止動(dòng)脈粥樣硬化的進(jìn)展.口服本品生物利用度高,半衰期長(zhǎng).具有肝臟選擇性,與通過細(xì)胞色素P450系統(tǒng)代謝的藥物發(fā)生相互作用的傾向更低.其降低甘油三酯的作用強(qiáng)于西立伐他汀、普伐他汀、氟伐他汀和辛伐他汀.臨床上本品用于治療高脂血癥.
安全信息
危險(xiǎn)運(yùn)輸編碼:暫無
危險(xiǎn)品標(biāo)志:暫無
安全標(biāo)識(shí):暫無
危險(xiǎn)標(biāo)識(shí):暫無
文獻(xiàn)
【1】 四川美康醫(yī)藥軟件研究開發(fā)有限公司編著.藥物臨床信息參考.成都:四川出版集團(tuán)四川科學(xué)技術(shù)出版社,2006:532. 【2】 Aoki T,et al.Arzneim-Forsch Drug Res,1997,47:904-909. 【3】 Miyachi N,et al.Tetrahedrom Lett,1993,34:8267-8270. 【4】 EP,304063.1989. 【5】 Wess G,et al.Tetrahedrom Lett,1990,31:2545-2548. 【6】 日本公開特許93-10700.(CA,1994,120:244704.) 【7】 EP,409281.1991. 【8】 蔡正艷等.中國醫(yī)藥工業(yè)雜志,2007,38:177-180. 【9】 Beck G,et al.Synthesis,1995,(8):1014-1018. 【10】 Manusco A J,et al.J Org Chem,1978,43:2480-2482. 【11】 Muller P,et al.Terachedrom Lett,1981,22:2361. 【12】 周偉澄主編.高等藥物化學(xué)選論.北京:化學(xué)工業(yè)出版社,2006:314-316. 【13】 Hiyama T,et al.Bull Chem Soc Jpn,1995,68:364-444.
備注
暫無
表征圖譜
暫無圖譜
統(tǒng)計(jì)數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競(jìng)化學(xué)品數(shù)據(jù)庫
-
物竟數(shù)據(jù)庫 手機(jī)版
國內(nèi)首款化工類專業(yè)手機(jī)應(yīng)用
-
微博賬號(hào)
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競(jìng)
物競(jìng)數(shù)據(jù)庫是一個(gè)全面、專業(yè)、專注,并且免費(fèi)的中文化學(xué)品信息庫,為學(xué)生、學(xué)者、化學(xué)品研究機(jī)構(gòu)、檢測(cè)機(jī)構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺(tái)進(jìn)行交流。
數(shù)據(jù)庫采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號(hào):物競(jìng)化學(xué)品數(shù)據(jù)庫

-
微博賬號(hào):wjhxp
聯(lián)系我們
上海市延長(zhǎng)路149號(hào)上海大學(xué)科技園412室
公司總機(jī): 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競(jìng)數(shù)據(jù)庫 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫 | 關(guān)于物競(jìng) | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號(hào)
滬公網(wǎng)安備 31010602001115號(hào)